肝豆状核变性研究的前世今生
2014年12月29日 8507人阅读 返回文章列表
肝豆状核变又称“Wilson’s病”,是遗传性铜代谢紊乱的常染色体隐性遗传病,主要累及中枢神经系统和肝脏。人们对肝豆状核变性的认识经历了从陌生到熟悉再到主动干预的过程。
昨天:阐明病理生理基础,发展驱铜治疗。
1912 年Wilson首次系统、详细地描述了肝豆状核变性的临床特征。1948年Cummings JN对于铜代谢紊乱的阐述使人们认识到铜排除障碍及其在肝脏、神经组织的沉积是肝豆状核变性的病理生理基础,各种驱铜药物应运而生,低铜饮食也因此成为重要的辅助治疗手段。1955年后青霉胺成为治疗该病的主流药物,同时,抑制铜吸收的锌剂、铜螯合剂曲恩汀、二巯基丙醇等也应用于该病的治疗。
驱铜药物的问世使人们能够主动控制疾病的发展,极大地改善了患者预后。然而,由于肝豆状核变性临床表现差异大、表现形式复杂、缺乏特异敏感的生化指标,因此常常误诊、漏诊而延误治疗。此外,驱铜药物的毒副作用较大,青霉胺初始治疗使20%~25%患者的病情加重,甚至发生不可逆恶化,长期治疗还可以引起免疫抑制、骨髓抑制、自身免疫疾病等,凸显了合理用药的重要性。
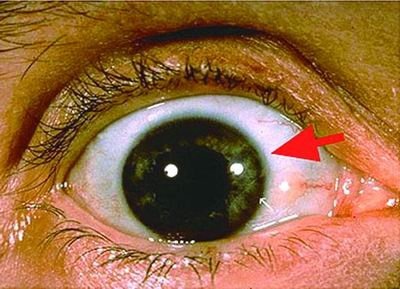
图 Wilson"s病眼部表现:箭头指示K-F环,即绿色的铜沉积在眼角膜周围,呈轮状带。
今天:分子水平研究进展迅速,治疗方面尚未突破。
带着如何提高早期诊断率、选择最佳治疗策略等问题,在分子生物学及医疗诊治技术飞速发展的大环境下,人们从对肝豆状核变性一知半解的“昨天”走进了更深层次认识、诊治疾病的“今天”。
我们对肝豆状核变性发病机制的认识已深入到了分子水平。1993年发现了位于13号染色体的ATP7B基因为该病的致病基因,其功能缺陷导致了肝脏铜的过度累积和肝脏损伤,当肝脏铜超负荷后,多余的铜又沉积在其他组织,主要是中枢神经系统,引起神经损害症状。肝豆状核变性是常染色体隐性遗传的疾病,因此患者的同胞有25%的患病几率,对明确诊断患者的家族进行基因筛查是对其他潜在患者进行早期诊断的最有效途径。ATP7B的变异位点繁多,存在显著的种族差异。例如:H1069Q变异在欧洲起源的白种人中最为常见,占37%~63%,却罕见于中国人群;中国人群中R778L是高频突变点,突变率达 28%~38%,而在印度人群中均未发现这两种变异,取而代之的是4193delC。因此,基因检测位点的选择要有种族特异性和针对性,避免无的放失。与患者拥有相同基因型的家族成员,即使没有临床症状也应尽早治疗;而临床症状或生化检查都模棱两可的家族成员,如果只是杂合子(携带者)或完全没有变异,则不必治疗。如果条件有限,不能进行基因鉴定,可以进行单倍体分析来确定是否具有与患者相同的两条同源染色体来判定患病机率。基因鉴定的方法不仅显著提高了不典型病例的诊断率,使肝豆状核变性发病年龄的确认被分别提前至3岁和延后至70岁,同时利用基因鉴定进行产前诊断也成为有效降低发病率的重要手段。
然而疾病的治疗水平并没有跟上“认识”的步伐,缺乏实质性的突破。治疗的核心仍以驱铜药物为主。由于缺乏大规模随机双盲对照的临床试验,迄今尚无公认的、有据可循的最优治疗方案,大多采取经验性治疗。虽然青霉胺仍是目前治疗肝豆状核变性的首选药物,但国内外学者开始更多关注曲恩汀,认为其疗效可比拟青霉胺而副作用更少,并推荐联用曲恩汀和锌剂作为初始治疗,适用于肝症状患者、神经精神症状患者以及尚处于临床前期的无症状患者。国内学者杨任民等研制“肝豆汤” 治疗肝豆状核变性总有效率达到84.85%,成为我国独特有效的治疗方法。
无论哪种治疗方案,都需要通过随访监测治疗效果、定期复诊来提高患者的依从性。然而仍有一部分患者虽经严格治疗,病情却持续恶化,肝脏损伤继续加重,符合肝移植适应证。由于肝移植需要长期免疫抑制剂治疗,并且生存期相对较短,因此所有准备接受肝移植的患者必须首先确认药物治疗无效。然而,对药物治疗成败的判定尚无定论。
在“今天”与肝豆状核变性的抗争中,人们的喜悦与挫折并存。尽管确定了疾病的遗传本质,克隆了其致病基因并发现了数百个变异位点,但仍有17%的确诊患者没有发现任何变异;在众多的变异位点和纷杂的临床表现间建立全面的基因型-表型关系,还需要做大量工作。尽管大多数患者经药物治疗有明显改善,但终生服药的难依从性影响了预后,单一的、局限的治疗手段远不能从根本上解决病因。以上种种困惑使我们无法满足于今天的成绩,而是面对更多的挑战。
图 肝豆状核变性的脑部病变
未来:治疗仍是最大难题
未来的几十年里,肝豆状核变性的研究将集中在以分子生物学手段为基础的诊断和治疗上,有遗传分析介入的诊断将更加重要。对患者的同胞进行基因检测有可能将诊断提前至无症状的临床前期,同时避免不必要的驱铜治疗。而ATP7B的外显子还有部分没有完成检测,有必要继续寻找新的突变位点,同时寻找更经济、快捷的基因鉴定法,使该项检查能够在所有高危人群中得到应用。
诊断明确后,如何在病因层次上进行治疗是“明天”面临的最严峻挑战。首先,为不同人群提供最佳治疗方案需要大量的循证医学证据,要求开展严格的临床对照试验。其次,肝豆状核变性的遗传特性使基因治疗成为根本的解决手段,这是由于患者肝脏解剖结构仍然正常,且肝脏本身具备适合基因治疗的特征。但基因治疗仍需克服重重难关,其中最重要的是建立安全有效的基因载体系统,后者应具备靶细胞特异性、可将遗传信息精确插入安全位点、能够接受生理信号调控、稳定表达以及方便给药等特点,建立这样的载体系统是基因治疗必须首先跨越的障碍。此外,通过正常肝细胞或干细胞移植来重建肝内铜代谢转运系统也前景光明,但细胞移植面临着供体细胞在宿主肝脏内的存活、增殖、避免瘤化、免疫排斥等问题。
一般而言,如果有30%的肝实质能够发挥正常生理功能就可以完成铜的代谢转运,并对其他肝细胞提供保护。因此无论是通过基因手段或是细胞移植方法,并不需要转染率达到100%或移植细胞全部存活,从而增强了临床应用的可能性。然而从实验研究到临床应用还需要相当长时间的摸索。
尽管尚有诸多障碍,我们已经踏上了彻底诊治肝豆状核变性的征途,昨天的无知结束于今天的探索,今天的挫折成为明天的起点,明天有挑战也有机遇,有困惑更有希望。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号