概述
多发内生软骨瘤病是1899年由Ollier首先描述,故亦称为Ollier病。它是一种少见的非遗传性良性肿瘤。常为多数的不对称的分布在骨内的软骨病灶及骨膜下沉积。在长、短管状骨中均可发病,可发生在肢体的单侧或双侧。
病因
是骨骼发育过程中部分异位的骨骺板衍变而成。
发病机制
肿瘤组织为白色,略有光泽,质脆,呈半透明状。掺杂黄色钙化或骨化区,或有黏液样退变区。显微镜下见分叶状透明软骨,软骨细胞成堆,有双核者,单核大小均匀,染色不深。
临床表现
通常发病年龄为10岁以内患儿,男性多于女性。 症状与体征:表现为可触及的肿块,但很少有疼痛。肿瘤侵及手部或足部,由于多发病变可以造成病残。病变侵及长管状骨,使内生软骨骨化不能正常进行,骨骺板不能正常生长,因而肢体可以出现短缩,弯曲畸形。如前臂向尺侧弯曲畸形,下肢膝外翻等。当患者达到成年时,肿瘤可停止生长。成人多发内生软骨瘤病可发生恶性变,恶变率约为5%~25%。 多发内生软骨瘤病,常在儿童时期出现症状,至青春期畸形明显,以后逐渐稳定。
并发症
可合并肢体短缩、弯曲畸形,例如膝外翻等。
实验室检查
无相关实验室检查。
其他辅助检查
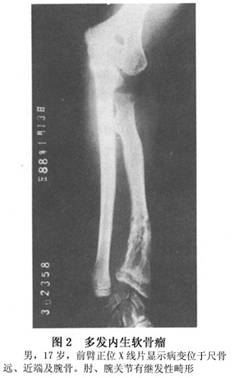
X线检查:多发内生软骨瘤病的每一个病变的X线表现与单发内生软骨瘤相似,但为多发。且有骨骼畸形或短缩(图1~3)。其干骺端可以增宽。

诊断
依据病史,临床症状及体征,X线的表现,一般诊断不难。
治疗
由于病变的多发性,难以将每个内生软骨瘤均予治疗,对无症状者可以不予治疗但应随诊观察。对有症状的具体部位,可以刮除病灶并植骨,对明显的肢体畸形可以做截骨纠正。 多发内生软骨瘤病有人报告可以随着生长病变可以缩小,甚至完全消失,而被正常组织所代替。但另一方面这种病变潜在性恶性改变的可能性较大,可变为软骨肉瘤或骨肉瘤,如有恶性变发生,则应采取较彻底手术方法予以切除,甚至截肢。
预后
多发内生软骨瘤具有潜在恶性变化的可能,恶变为软骨肉瘤或骨肉瘤,应引起注意。
预防
无相关资料。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号