
概述
神经纤维瘤病(neurofibromatosis,NF-1)亦属斑痣性错构瘤病之一。1882年Von Recklinghausen首先报告本病的临床和病理改变,所以又名Recklinghausen病、von Recklinghausen病。是遗传性疾患,属常染色体显性遗传,外显率不规则,遗传特性的表现为多变性,突变率高。发病率约为新生儿的1/3000,发病年龄可在出生时、儿童后期或成人。本病原发生于神经外胚叶组织细胞的生长发育障碍,其特征是周围神经纤维增殖而形成肿瘤样结节,侵入皮肤、内脏、神经系统,并伴有皮肤色素沉着斑。临床以皮肤色素异常斑和躯干、四肢以及眼部周围神经多发性肿瘤样增生为特点,又...[详细]
病因
神经纤维瘤病多属于一种常染色体显性遗传病,但也有隐性遗传病例报告。神经外胚叶组织细胞的生长发育障碍。
发病机制
目前对其发病机制尚未完全了解。有些人认为神经纤维瘤发病与胚胎时期神经嵴细胞的发育异常有关,也有人认为其发病原因主要是因为细胞间的相互作用与细胞外环境的化学性质共同影响所致。还有人认为神经生长素的分泌与此病的发病相关。 关于青光眼的发病机制,一般认为房水流出受阻的主要机制是神经纤维瘤直接侵犯房角,可见一层无血管的透明的致密组织从周边虹膜向前扩展覆盖在房角壁上。另一种因素为睫状体与脉络膜受神经纤维瘤的累及而变肥厚,向前推移使房角关闭。部分病例有前房角发育不良、房角胚胎组织残留、房角分裂不全、Schlemm管畸形或残缺等。房角及虹膜根部直接被神经纤维瘤侵犯时可导致虹膜广泛前粘连,形成纤维血...[详细]
临床表现
多数神经纤维瘤是神经纤维瘤病的表现之一,而神经纤维瘤病有多种临床表现,可累及中枢神经系统、周围神经系统、骨骼、肌肉、皮肤和眼部。眼部常是神经纤维瘤病的好发部位,几乎可侵及所有眼部结构和组织。主要表现: 1.眼部表现 眼部易累及的部位依次为眼睑、眼眶、葡萄膜、视神经、角膜、结膜、巩膜,晶状体和玻璃体一般不受累。 (1)眼睑:眼睑是最常受累的部位,上睑多见,常为单侧性。上眼睑的丛状神经纤维瘤睑皮下瘤细胞弥漫性增生,软性肥厚无边界,如面团状,会引起机械性上睑下垂,呈特征性的“S”状上眼睑畸形。病变不断增生引起睑裂延长,上睑外翻,也可有双上睑及双下睑发病。此丛状...[详细]

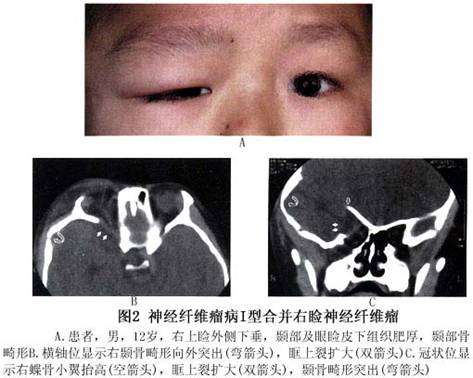
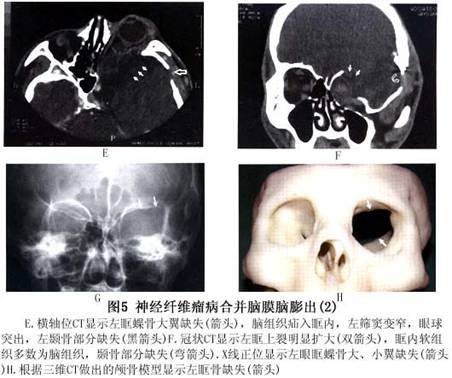
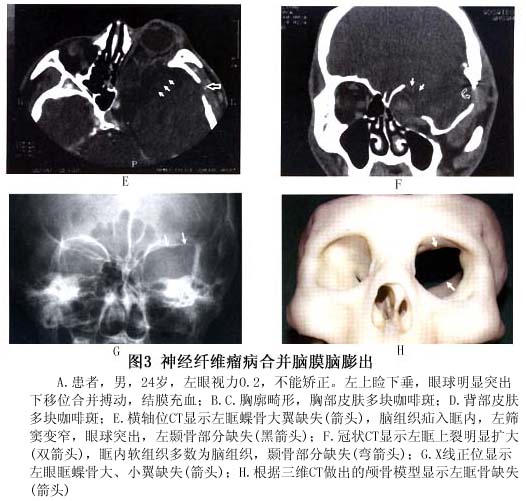 (2)眼眶:眼眶内结节状或丛状神经纤维瘤:可使眼球突出(图4,5),眶壁、蝶骨发育不良,常显示眶外壁、眶顶骨质缺失,眶上裂扩大,范围较大的骨缺损导致脑膜或脑膜脑组织疝入眶内,出现搏动性眼球突出(图4,5),因系由颈动脉搏动,通过颅内传导而来,故无血管杂音,扪诊也无震颤。
(2)眼眶:眼眶内结节状或丛状神经纤维瘤:可使眼球突出(图4,5),眶壁、蝶骨发育不良,常显示眶外壁、眶顶骨质缺失,眶上裂扩大,范围较大的骨缺损导致脑膜或脑膜脑组织疝入眶内,出现搏动性眼球突出(图4,5),因系由颈动脉搏动,通过颅内传导而来,故无血管杂音,扪诊也无震颤。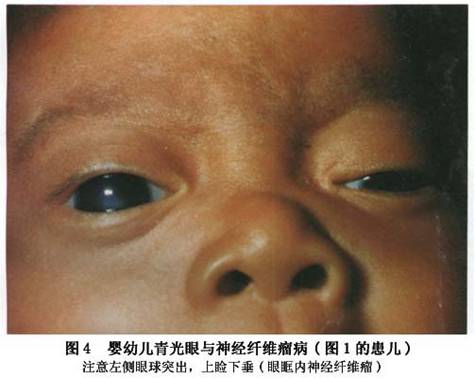
 (3)虹膜错构瘤(Lisch结节):虹膜错构瘤常见于双侧性,呈半球形白色或黄棕色隆起斑点,境界清楚的胶样结节,称为虹膜结节。隆起于虹膜面(图6)。常从16岁开始生长,随年龄增大而继续发展。另外还可以有先天性葡萄膜外翻、虹膜异色的存在。
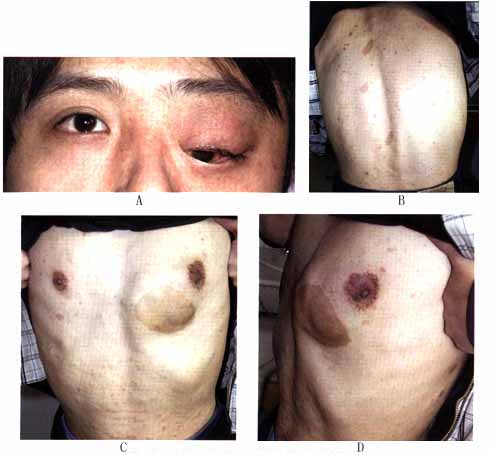
(3)虹膜错构瘤(Lisch结节):虹膜错构瘤常见于双侧性,呈半球形白色或黄棕色隆起斑点,境界清楚的胶样结节,称为虹膜结节。隆起于虹膜面(图6)。常从16岁开始生长,随年龄增大而继续发展。另外还可以有先天性葡萄膜外翻、虹膜异色的存在。 (4)脉络膜错构瘤:发生率为30%,其呈棕黑色扁平状或轻度隆起,散在性地分布于多色素的区域。 (5)视神经:可发生胶质瘤或错构瘤,约有1/4~1/2视神经胶质瘤患者合并神经纤维瘤病。也可以是真性肿瘤。瘤体常在蛛网膜下隙内增殖,4~8岁时起病,表现为单侧眼球进行性突出和视力丧失、视盘水肿或萎缩。90%的病例肿瘤会累及视神经管的前段而引起视神经孔扩大,B型超声波或CT检查可见视神经肿大,X线显示视神经孔扩大。 (6)视网膜:亦可发生胶质错构瘤,角膜神经粗大,结膜、浅层巩膜偶有纤维增生或肿物,巩膜可有色素沉着,有时可引起眼球增大呈牛眼状,但眼压并不高。 (7)神经纤维瘤合并青光眼:可因前房角有异常组织阻塞致先天性青光眼,眼球直径较大。当肿物累及同侧上眼睑或眼球本身时,则应注意可能已经合并有青光眼,其发病率可高达50%。青光眼常在出生时或出生后不久发生,也偶有晚发者。临床上多为开角型,单眼居多。如青光眼较早发生,则出现先天性青光眼的各种病变,如发生较晚则与成年人的开角型青光眼相似。 2.全身表现 (1)皮肤咖啡样色素斑:这是本病最常见的体征,又称为Cafeau Lait斑,其呈多发性,大小不等(数毫米至数厘米),边缘不规则的淡棕色斑,多位于躯干部,位于背部、腋下,其数目常与疾病严重程度有关。只要发现一处,就应考虑到神经纤维瘤病。这些皮肤咖啡斑在6个以上,直径在15mm或一个直径在6cm以上有诊断意义。此斑在出生时或出生后不久即已出现,在儿童期此斑的大小和数量会增多。病理改变为多巴阳性色素细胞的增多引起基底细胞层色素的增生所致。 (2)皮肤神经纤维瘤:周围神经鞘细胞增生而形成的弥漫性丛状神经瘤。其在青春期出现,在一生中瘤体的数量会增多。因为瘤所在的表面皮肤变厚、起褶,触诊时感觉似一个虫袋,所以又称为神经瘤性象皮病。有的表现为纤维软疣,为具有色素和带蒂的、松软的瘤结节,由结缔组织、增生的神经鞘细胞和增大的皮神经组成。这些皮下肿物为多发性,严重病例可有数百个,遍布全身,大小不一,小如豌豆,大如鸡蛋,多沿神经干分布,呈念珠状,大多数突出体表,有的在皮下(图7)。皮肤或皮下神经纤维瘤是本病主要诊断指征。根据国际诊断标准,以上所述为Ⅰ型神经纤维瘤病(NF-1)。临床上还有一种特殊类型是神经纤维瘤病Ⅱ型。国际上区别神经纤维瘤病I和Ⅱ型标准如下:
(4)脉络膜错构瘤:发生率为30%,其呈棕黑色扁平状或轻度隆起,散在性地分布于多色素的区域。 (5)视神经:可发生胶质瘤或错构瘤,约有1/4~1/2视神经胶质瘤患者合并神经纤维瘤病。也可以是真性肿瘤。瘤体常在蛛网膜下隙内增殖,4~8岁时起病,表现为单侧眼球进行性突出和视力丧失、视盘水肿或萎缩。90%的病例肿瘤会累及视神经管的前段而引起视神经孔扩大,B型超声波或CT检查可见视神经肿大,X线显示视神经孔扩大。 (6)视网膜:亦可发生胶质错构瘤,角膜神经粗大,结膜、浅层巩膜偶有纤维增生或肿物,巩膜可有色素沉着,有时可引起眼球增大呈牛眼状,但眼压并不高。 (7)神经纤维瘤合并青光眼:可因前房角有异常组织阻塞致先天性青光眼,眼球直径较大。当肿物累及同侧上眼睑或眼球本身时,则应注意可能已经合并有青光眼,其发病率可高达50%。青光眼常在出生时或出生后不久发生,也偶有晚发者。临床上多为开角型,单眼居多。如青光眼较早发生,则出现先天性青光眼的各种病变,如发生较晚则与成年人的开角型青光眼相似。 2.全身表现 (1)皮肤咖啡样色素斑:这是本病最常见的体征,又称为Cafeau Lait斑,其呈多发性,大小不等(数毫米至数厘米),边缘不规则的淡棕色斑,多位于躯干部,位于背部、腋下,其数目常与疾病严重程度有关。只要发现一处,就应考虑到神经纤维瘤病。这些皮肤咖啡斑在6个以上,直径在15mm或一个直径在6cm以上有诊断意义。此斑在出生时或出生后不久即已出现,在儿童期此斑的大小和数量会增多。病理改变为多巴阳性色素细胞的增多引起基底细胞层色素的增生所致。 (2)皮肤神经纤维瘤:周围神经鞘细胞增生而形成的弥漫性丛状神经瘤。其在青春期出现,在一生中瘤体的数量会增多。因为瘤所在的表面皮肤变厚、起褶,触诊时感觉似一个虫袋,所以又称为神经瘤性象皮病。有的表现为纤维软疣,为具有色素和带蒂的、松软的瘤结节,由结缔组织、增生的神经鞘细胞和增大的皮神经组成。这些皮下肿物为多发性,严重病例可有数百个,遍布全身,大小不一,小如豌豆,大如鸡蛋,多沿神经干分布,呈念珠状,大多数突出体表,有的在皮下(图7)。皮肤或皮下神经纤维瘤是本病主要诊断指征。根据国际诊断标准,以上所述为Ⅰ型神经纤维瘤病(NF-1)。临床上还有一种特殊类型是神经纤维瘤病Ⅱ型。国际上区别神经纤维瘤病I和Ⅱ型标准如下: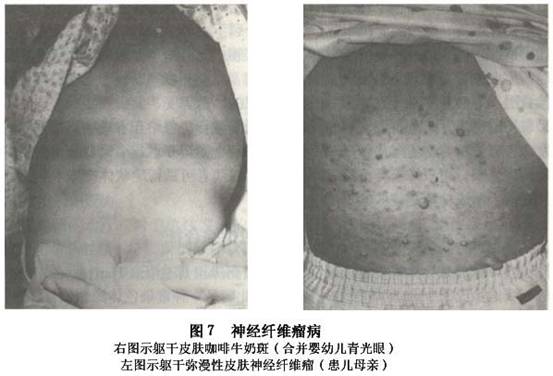 ①神经纤维瘤病Ⅰ型:是一种常染色体遗传性疾病,发病率约1/3000。其基因位于17号染色体长臂。Ⅰ型神经纤维瘤病诊断标准: 有下述两个以上症状者,可以诊断神经纤维瘤病Ⅰ型: A.6个以上咖啡斑,青春期前直径>6cm,青春期后直径>15cm。 B.两个以上任何类型的神经纤维瘤或一个丛状神经纤维瘤。 C.腹部或纵轴区的雀斑。 D.明确的骨性病变,如蝶骨发育异常或骨皮质变薄合并或不合并假关节病。 E.视神经胶质瘤。 F.2个以上的Lisch结节(虹膜错构瘤)。 G.父母、同胞之一患有神经纤维瘤病Ⅰ型或具有神经纤维瘤病Ⅰ型的子女。 ②神经纤维瘤病Ⅱ型(NF-2):以前称双侧听神经瘤神经纤维瘤病(bilateral acoustic neurofibromatosis)或中枢神经纤维瘤病(central neurofibromatosis),是临床较少见的常染色体畸形,发病率约1/50000~1/40000,基因位于22号常染色体。Ⅱ型神经纤维瘤病诊断标准: 有以下两条之一者: A.增强MRI或CT扫描证实双侧听神经瘤。 B.父母、同胞之一患有神经纤维瘤病Ⅱ型或具有神经纤维瘤病Ⅱ型的子女,或有单侧听神经瘤或具有下列任何体征之一:a.神经纤维瘤。b.脑膜瘤。c.胶质瘤。d.神经鞘瘤。e.年轻患者的后囊下白内障。 (3)骨骼系统的改变:约有29%的患者有先天性骨骼缺陷,表现为骨质肥大及侵蚀,X线及显微镜下所见同囊性纤维性骨炎相似,常累及蝶骨和眼眶,脊柱和肢体也可受累。 (4)神经系统改变:中枢神经系统受累的同时,常有颅内肿物,如脑膜瘤、胶质瘤等。肿瘤可引起颅内高压、眩晕、运动或知觉障碍、视野缺损。双侧听神经瘤较多见,产生脑桥角症状。另外,脊髓、脑神经、周围神经、交感神经和肾上腺(嗜铬细胞瘤)等均可累及。 (5)其他:部分患者有半侧面部萎缩,少数可有智力低下、精神障碍、隐睾等。
①神经纤维瘤病Ⅰ型:是一种常染色体遗传性疾病,发病率约1/3000。其基因位于17号染色体长臂。Ⅰ型神经纤维瘤病诊断标准: 有下述两个以上症状者,可以诊断神经纤维瘤病Ⅰ型: A.6个以上咖啡斑,青春期前直径>6cm,青春期后直径>15cm。 B.两个以上任何类型的神经纤维瘤或一个丛状神经纤维瘤。 C.腹部或纵轴区的雀斑。 D.明确的骨性病变,如蝶骨发育异常或骨皮质变薄合并或不合并假关节病。 E.视神经胶质瘤。 F.2个以上的Lisch结节(虹膜错构瘤)。 G.父母、同胞之一患有神经纤维瘤病Ⅰ型或具有神经纤维瘤病Ⅰ型的子女。 ②神经纤维瘤病Ⅱ型(NF-2):以前称双侧听神经瘤神经纤维瘤病(bilateral acoustic neurofibromatosis)或中枢神经纤维瘤病(central neurofibromatosis),是临床较少见的常染色体畸形,发病率约1/50000~1/40000,基因位于22号常染色体。Ⅱ型神经纤维瘤病诊断标准: 有以下两条之一者: A.增强MRI或CT扫描证实双侧听神经瘤。 B.父母、同胞之一患有神经纤维瘤病Ⅱ型或具有神经纤维瘤病Ⅱ型的子女,或有单侧听神经瘤或具有下列任何体征之一:a.神经纤维瘤。b.脑膜瘤。c.胶质瘤。d.神经鞘瘤。e.年轻患者的后囊下白内障。 (3)骨骼系统的改变:约有29%的患者有先天性骨骼缺陷,表现为骨质肥大及侵蚀,X线及显微镜下所见同囊性纤维性骨炎相似,常累及蝶骨和眼眶,脊柱和肢体也可受累。 (4)神经系统改变:中枢神经系统受累的同时,常有颅内肿物,如脑膜瘤、胶质瘤等。肿瘤可引起颅内高压、眩晕、运动或知觉障碍、视野缺损。双侧听神经瘤较多见,产生脑桥角症状。另外,脊髓、脑神经、周围神经、交感神经和肾上腺(嗜铬细胞瘤)等均可累及。 (5)其他:部分患者有半侧面部萎缩,少数可有智力低下、精神障碍、隐睾等。并发症
神经纤维瘤病可合并眼眶神经鞘瘤、脑膜瘤、视神经和视交叉胶质瘤,定期复查MRI对早期发现、早期诊断和治疗非常重要。
实验室检查
1.遗传学检查 开展一些必要的遗传学检查,基因分析可确定NFⅠ和NFⅡ突变类型。 2.病理学检查 主要特征是神经外胚层结构过度增生和肿瘤形成,尚伴有中胚层组织过度增生,典型病理改变为梭形细胞组成的神经纤维瘤、大小不一,主要生长于周围神经,也可发生在颅神经、脊神经或马尾等处,神经纤维瘤基本由鞘膜细胞组成。有些学者认为肿瘤是周围Schwann细胞弥漫增生的结果,另一些学者则认为来源于神经内膜和外膜。
其他辅助检查
1.X线检查 对神经纤维瘤病(NF-1)的诊断主要依靠眶周围骨质缺失、眶内和颞部皮下软组织增厚或条索状软组织块影。有无先天性骨骼缺陷,表现为骨质肥大及侵蚀,类似纤维性囊性骨炎,特别注意蝶骨大翼和眼眶。 2.B超或CT可明确视神经及视网膜情况,X线检查视神经孔情况。 3.出现青光眼时注意眼压的监测。 4.CT和 MRI检查 NF-1神经系统病变主要包括视路胶质瘤、非新生物性错构瘤、胶质瘤、丛状神经纤维瘤、散在脊椎内神经纤维瘤和硬膜扩张症。各种结构上的异常也常发现,如巨头症(macrocephaly)。仅用MRI上显示的儿童大脑内许多错构瘤或高信号性病变即可诊断N...[详细]
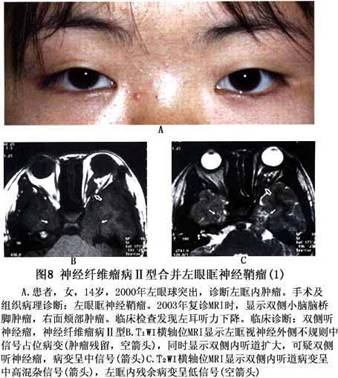
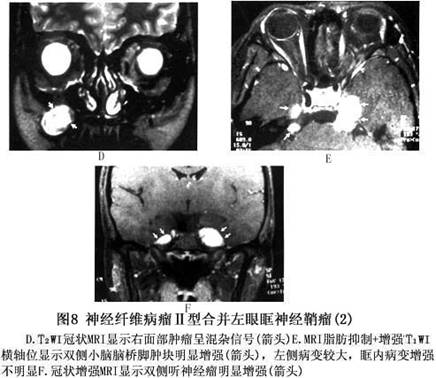
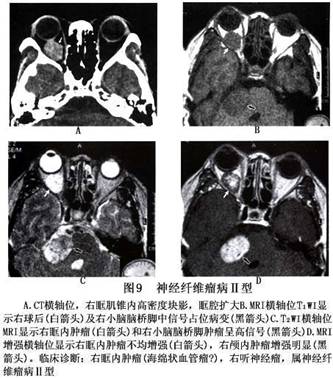
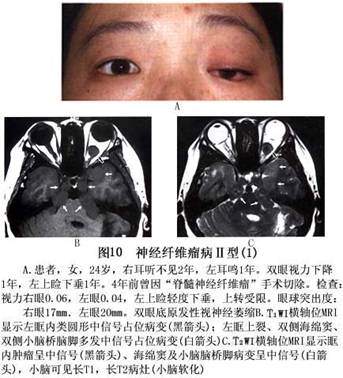
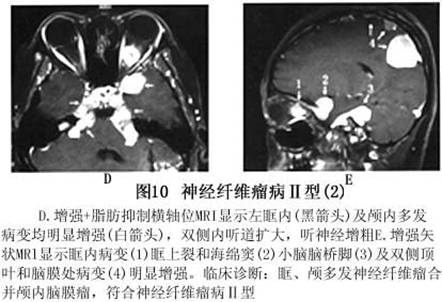
诊断
根据该病的典型的临床表现,并结合辅助检查可以明确诊断。神经纤维瘤病的诊断标准有: 1.皮肤色素斑6个或6个以上,直径至少15mm或腋窝雀斑。 2.皮肤和皮下良性神经纤维瘤是本病主要标志。 3.“中枢型”在缺乏皮肤体征时诊断较困难。可根据有无阳性家族史、双侧听神经瘤、单侧搏动性眼球突出,以及智力迟钝、癫痫发作、脊柱畸形等发现,并经仔细检查,在获得皮肤体征时可确诊。 4.对少数疑难病例,需经活检或病程达一定阶段时才能确诊。 神经纤维瘤病严重性分级: Ⅰ级:最轻度(皮肤表现、色素斑、无并发神经纤维瘤)。 Ⅱ级:轻度(轻度脊柱侧曲,...[详细]
治疗
症状和外观不很明显的神经纤维瘤病可不予治疗。本病并发青光眼时,可根据青光眼的严重程度、发病年龄及不同的发病机制,采取相应措施,儿童早期发病的开角型青光眼可行房角切开术或小梁切开术,儿童后期发病者可先用药物治疗,如疗效不佳,再采用小梁切开术或小梁切除术。对房角已经关闭的青光眼,则只好施行小梁切除术,若不成功者可试行睫状体冷凝术。此种继发性青光眼的手术成功率是较低的。 1.眼睑丛状神经纤维瘤明显者可行病变切除,眼睑整形术,但复发率较高,部分病例数年内需要再次手术。 2.合并眶内病变可手术切除。 3.神经纤维瘤病Ⅱ型根据情况处理,尤其是单侧听神经瘤如体积不大,可考虑伽玛...[详细]
预后
手术成功率低,预后不良。本病预后与症状轻重程度有关,有肿瘤引起颅内及内脏病变者,预后较差。
预防
目前无相关内容。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号



