概述
在婴幼儿眼病中,是性质最严重、危害性最大的一种恶性肿瘤视网膜母细胞瘤(retinoblastoma,RB)是婴幼儿最常见的眼内恶性肿瘤,对视力和生命有严重的威胁和危害。发生于视网膜核层,具有家族遗传倾向,多发生于5岁以下,可单眼、双眼先后或同时罹患,本病易发生颅内及远处转移,常危及患儿生命,因此早期发现、早期诊断及早期治疗是提高治愈率、降低死亡率的关键。
病因
遗传学类型及Rb基因的定位:90%的视网膜母细胞瘤(包括单眼和双眼病例)表现为散发发病,10%的病例为家族性发病。视网膜母细胞瘤可分为遗传型和非遗传型2大类,其发生有3种情况:①约40%的病例属于遗传型,是由患病或基因携带者父母遗传所致,或正常父母生殖细胞突变所致,为常染色体显性遗传。这类患者发病早,约85%为双眼发病,有多个病灶,易发生第2恶性肿瘤。约15%为单眼发病,其原因可能是视网膜母细胞瘤基因外显不全。一般公认本病外显率为90%左右。临床上将双眼视网膜母细胞瘤、有家族史的单眼视网膜母细胞瘤或多病灶的单眼视网膜母细胞瘤归入遗传型。②约60%的病例属于非遗传型,其发病系患者视网膜母细胞发生...[详细]
发病机制
1.Rb基因突变 Rb基因是人类发现的第1个肿瘤抑制基因,Rb基因的发现被公认为人类肿瘤学研究、细胞周期研究的一个重要里程碑。Rb基因定位于13q14,全长约180kb,共27个外显子,转录成1条长4.7kb的mRNA,编码具有928个氨基酸残基的Rb蛋白。大约80%的视网膜母细胞瘤可发现Rb基因突变,主要有4种类型:无功能(null)突变、阅读框架内(in frame)突变、启动子突变(点突变和甲基化)和LOH。阅读框架内突变的Rb基因仍有部分正常功能。早期Rb基因突变的检测主要靠Southern杂交,目前主要用定量PCR及直接DNA测序。通过对比患者肿瘤及外周血白细胞的Rb基因突变状态可...[详细]
临床表现
1.根据视网膜母细胞瘤一般的发展过程,临床可分为4期,即眼内生长期、眼压增高期(青光眼期)、眼外扩展期及全身转移期。由于肿瘤生长部位、生长速度和分化程度不同,临床表现也不尽一致。例如生长在视盘附近或视网膜周边部的肿瘤,可早期侵犯视神经或睫状体向眼外转移,并不经过青光眼期而直接进入眼外扩展期;又如临床上诊断为青光眼期者,病理学检查已可能有眼外扩展。 (1)眼内生长期:其早期症状和体征是视力障碍和眼底改变。早期病变可发生于眼底任何部位,但以后极部偏下方为多。若肿瘤发生于视网膜内核层,易向玻璃体内生长,称为内生型。眼底检查可见肿瘤呈圆形或椭圆形,边界不清,呈白色或黄白色的结节,表面有新生血...[详细]
并发症
肿瘤发展的不同时期可产生多种不同的并发症。包括玻璃体混浊、视网膜脱离、新生血管性青光眼等。
实验室检查
1.尿液检查 尿液中香草基苦杏仁酸(vanilmandelic acid)和高香草基酸(homovanillic acid)排出量增加。阳性者有助诊断。但阴性者亦不能排除Rb。 2.在血-房水屏障完整时,房水中乳酸脱氢酶(LDH)浓度高于血清值。当两者之比值大于1.5时,提示Rb存在的可能。 3.房水酶的测定 视网膜母细胞瘤患者房水及血浆的乳酸脱氢酶(LDH)增高,房水与血浆的磷酸异构酶(PGI)亦增高,但是在Coats病晚期,视网膜破坏较广时,房水和血浆的乳酸脱氢酶及磷酸异构酶亦增高。 4.细胞学检查 抽取房水或玻璃体进行细胞学检查,对于本病的诊断和鉴别诊...[详细]
其他辅助检查
1.眼眶X线相片 视网膜母细胞瘤在眼眶X相片上可显示出不正常的钙化。 2.超声探查 当患儿因斜视或“猫眼”就诊时,瘤体一般较大,超声检查有典型的表现,对诊断有重要的意义。肿瘤常有钙化,表现为高反射伴声影。少数肿瘤因生长过快,出现液化、坏死而无钙化,为低反射。眼球可正常或增大,测量眼轴可鉴别眼轴短的白瞳症(原始玻璃体组织增生症)。肿瘤可单个或多灶。弥漫型者较少,表面轮廓不规则,无钙化。检查时因患儿不合作,常使用镇静剂。应双眼检查,必要时需重复检查。 视网膜母细胞瘤(有钙化)的超声诊断依据(图1,2): A型:①极高的反射(坏死液化区为超低的反射);②单个...[详细]
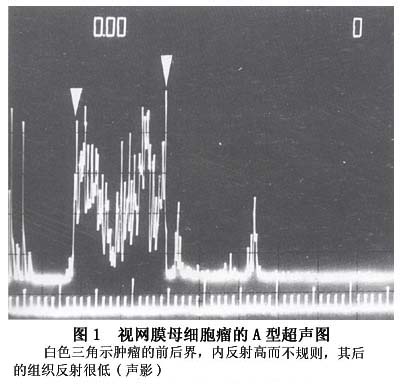
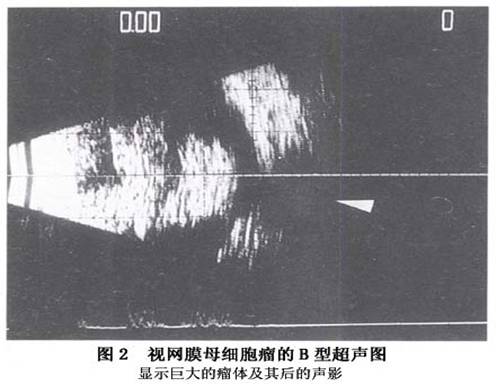 A型:①极高的反射(坏死液化区为超低的反射);②单个回声源;③有声影。B型:①视网膜组织破坏;②形状不规则的肿块;③有声影。 3.电子计算机体层扫描(CT扫描)及磁共振(MRI扫描) CT和MRI扫描不仅可发现和描画出肿瘤的位置、形状和大小,而且可查出肿瘤向眼球外蔓延引起的视神经粗大、眶内包块及颅内转移等情况。CT扫描也可显示肿瘤内的钙化,对诊断极有参考价值。 4.眼底图像采集 定期对眼底肿瘤进行照相、摄像,有助于诊断及病情判断,可很好地指导治疗。目前被广泛应用的是一种眼底广角摄像机RETCAM,可在术中应用。 5.肉眼分型 一般肉眼下即可见到视网膜内黄白色的肿瘤,常常可见到致密的钙化灶。根据肉眼观察,视网膜母细胞瘤有3种类型:①内生型:肿瘤起源于视网膜内核层,向玻璃体内生长,早期易为眼底检查所发现;②外生型:肿瘤起源于视网膜外核层,沿视网膜下间隙及脉络膜方向生长,造成视网膜脱离,检眼镜早期不易发现肿瘤团块。③浸润型:肿瘤弥散性的浸润视网膜全层,无明显包块。占全部视网膜母细胞瘤的1.5%,多见于非遗传型视网膜母细胞瘤。
A型:①极高的反射(坏死液化区为超低的反射);②单个回声源;③有声影。B型:①视网膜组织破坏;②形状不规则的肿块;③有声影。 3.电子计算机体层扫描(CT扫描)及磁共振(MRI扫描) CT和MRI扫描不仅可发现和描画出肿瘤的位置、形状和大小,而且可查出肿瘤向眼球外蔓延引起的视神经粗大、眶内包块及颅内转移等情况。CT扫描也可显示肿瘤内的钙化,对诊断极有参考价值。 4.眼底图像采集 定期对眼底肿瘤进行照相、摄像,有助于诊断及病情判断,可很好地指导治疗。目前被广泛应用的是一种眼底广角摄像机RETCAM,可在术中应用。 5.肉眼分型 一般肉眼下即可见到视网膜内黄白色的肿瘤,常常可见到致密的钙化灶。根据肉眼观察,视网膜母细胞瘤有3种类型:①内生型:肿瘤起源于视网膜内核层,向玻璃体内生长,早期易为眼底检查所发现;②外生型:肿瘤起源于视网膜外核层,沿视网膜下间隙及脉络膜方向生长,造成视网膜脱离,检眼镜早期不易发现肿瘤团块。③浸润型:肿瘤弥散性的浸润视网膜全层,无明显包块。占全部视网膜母细胞瘤的1.5%,多见于非遗传型视网膜母细胞瘤。诊断
根据临床表现并结合辅助检查结果,容易确定诊断。
治疗
视网膜母细胞瘤作为一发生在儿童的遗传性眼内恶性肿瘤,影响患儿生命、视力、面部外形及心理发育。诊断上涉及眼科、儿科、产科,治疗上涉及眼科、肿瘤科(放疗、化疗)、儿科、麻醉科等。因此在对视网膜母细胞瘤的处理上,一定要强调多学科、多中心的合作。视网膜母细胞瘤的治疗目标首先是挽救生命,其次是保留眼球及部分视力。治疗原则应根据眼部及全身受肿瘤侵犯的情况而定。方法的选择应根据肿瘤的大小和范围,单侧或双侧以及患者的全身情况而定。 常用的治疗方法有手术治疗(包括眼球摘除、眼眶内容摘除)、外部放射治疗、局部治疗(光凝治疗、冷冻治疗、加热治疗、浅层巩膜贴敷放射治疗)及化学治疗等。近10年来国际上对视网膜...[详细]
预后
1.生命预后 近200年来,视网膜母细胞瘤的生命预后已有很大改善。1个世纪前病死率为100%,由于诊断和治疗技术的改进,目前在欧美及其他工业国家,本病病死率已下降到5%以下。生命预后与许多因素有关,如肿瘤的大小和部位,诊断和治疗的迟早,治疗措施是否合理等。预后亦与组织学改变有关,一般来说,分化程度好的较分化程度低的预后好;肿瘤限于视网膜者较侵犯脉络膜、视神经或已有眼外扩散者好。死因分析,50%的患者死于肿瘤的眼外转移,50%是由于发生了第2恶性肿瘤。 2.视力预后 单眼患者未受累眼的视力预后是良好的。在患眼摘除或治疗后,另眼应定期检查,多数患儿成年后,健眼视力良好。双眼患者视力预...[详细]
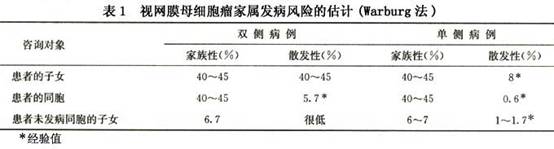 (2)通过对Rb基因突变的检测来进行遗传咨询:DNA样本可取自外周血白细胞和视网膜母细胞瘤瘤组织。多种基因突变检测技术如Southern杂交、SSCP、DGGE等,基因剂量检测技术如定量PCR,直接DNA测序均可应用。由于费用、时间的限制临床上不可能对Rb基因全长180kb的序列进行全部检测,一般集中在27个外显子和外显子附近10~20bp的内含子序列上(共约4kb)。Rb基因突变类型可是整个基因的缺失,也可小至点突变。一般可在瘤组织中发现两个突变(可相同也可不同),如果在外周血白细胞中也存在其中的一个突变则可判断为遗传型视网膜母细胞瘤,如果在外周血白细胞中不存在突变则可判断为非遗传型视网膜母细胞瘤。对遗传型视网膜母细胞瘤患者的亲戚可采血检查是否有相同的Rb基因突变,若有此突变则其本人及子女有90%患病的风险,若无则风险较低。在用DNA检查进行遗传咨询时要注意嵌合(mosaic)和低外显率(low penetrance)现象。 3.产前诊断 Rb基因突变检测已成功应用于临床产前诊断。对于遗传型视网膜母细胞瘤家族的胎儿可于妊娠28~30周取羊水细胞作Rb基因突变检测,若存在该家族的Rb基因突变,最好终止妊娠;若胎儿父母不愿终止妊娠,可于妊娠33~35周行经阴道的B超检查,每周1~2次,观察胎儿眼内是否形成肿瘤,若肿瘤已形成可在妊娠35周引产,立即对肿瘤进行激光治疗。有报告经上述妊娠35周引产及激光治疗的视网膜母细胞瘤,最终不仅保留了眼球,也保留了良好的视力。
(2)通过对Rb基因突变的检测来进行遗传咨询:DNA样本可取自外周血白细胞和视网膜母细胞瘤瘤组织。多种基因突变检测技术如Southern杂交、SSCP、DGGE等,基因剂量检测技术如定量PCR,直接DNA测序均可应用。由于费用、时间的限制临床上不可能对Rb基因全长180kb的序列进行全部检测,一般集中在27个外显子和外显子附近10~20bp的内含子序列上(共约4kb)。Rb基因突变类型可是整个基因的缺失,也可小至点突变。一般可在瘤组织中发现两个突变(可相同也可不同),如果在外周血白细胞中也存在其中的一个突变则可判断为遗传型视网膜母细胞瘤,如果在外周血白细胞中不存在突变则可判断为非遗传型视网膜母细胞瘤。对遗传型视网膜母细胞瘤患者的亲戚可采血检查是否有相同的Rb基因突变,若有此突变则其本人及子女有90%患病的风险,若无则风险较低。在用DNA检查进行遗传咨询时要注意嵌合(mosaic)和低外显率(low penetrance)现象。 3.产前诊断 Rb基因突变检测已成功应用于临床产前诊断。对于遗传型视网膜母细胞瘤家族的胎儿可于妊娠28~30周取羊水细胞作Rb基因突变检测,若存在该家族的Rb基因突变,最好终止妊娠;若胎儿父母不愿终止妊娠,可于妊娠33~35周行经阴道的B超检查,每周1~2次,观察胎儿眼内是否形成肿瘤,若肿瘤已形成可在妊娠35周引产,立即对肿瘤进行激光治疗。有报告经上述妊娠35周引产及激光治疗的视网膜母细胞瘤,最终不仅保留了眼球,也保留了良好的视力。 浙公网安备
33010902000463号
浙公网安备
33010902000463号