概述
假性醛固酮减少症(pseudohypoaldosteronism)又称Cheek-Perry综合征,系Cheek及Perry(1958)首次报道,是一种少见的失盐综合征。目前认为本病的病因是病人的靶器官(肾小管、唾液腺、汗腺和结肠)上的醛固酮受体缺乏,或醛固酮与其受体结合减少或完全不能结合所致;分子生物学及分子生物化学的进一步研究发现假性醛固酮减少症的病因学基础是由基因决定的细胞膜上钠通道功能障碍。在不同的病人受体的靶器官不一定相同,其临床表现的失盐程度也轻重不一。由于大多数病人可追溯出失盐的家族史,因此,有报道认为本病是一种遗传性疾病,其遗传方式可表现为常染色体显性遗传,也可表现为常染色体隐...[详细]
病因
本病是一种遗传性疾病,其遗传方式可表现为常染色体显性遗传,也可表现为常染色体隐性遗传。
发病机制
假性醛固酮减少症可分为Ⅰ型与Ⅱ型,其中Ⅰ型呈常染色体隐性遗传者,基因突变使集合管上皮细胞钠通道蛋白亚单位功能丧失,钠再吸收功能障碍。肾小管细胞对内源性醛固酮反应性减低,肾小球及其他肾小管功能正常。常色体显性遗传者为盐皮质类固醇受体功能障碍Ⅱ型,发病机制不明。Ⅰ型发病机制如图1。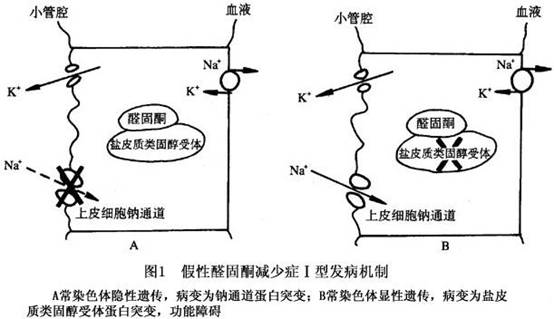
临床表现
发病年龄多在新生儿期,可于生后数小时出现症状反复呕吐、腹泻,渴感减退或消失,生长发育落后为主要症状(甚至是白痴);有些病例则于限盐或应用醛固酮拮抗剂才显露症状,并随年龄增长而自行缓解。血液生化改变为低钠、低氯和高钾血症,部分病人可有酸中毒;同时存在有高肾素血症和高血浆醛固酮活性。尿中醛固酮排量增大,但尿17酮及17羟类固醇及ACTH试验正常;使用外源性醋酸脱氧皮质酮或9α-氟氢可的松无反应。患儿多因高尿钠引起多尿、低渗或等渗性脱水、严重电解质紊乱,如未及时采取有效治疗,多因脱水或继发感染而夭折。
并发症
生长发育落后;电解质紊乱酸中毒;继发各种感染性疾病。
实验室检查
血液生化改变为低钠、低氯和高钾血症,部分病人可有酸中毒;同时存在有高肾素血症和高血浆醛固酮活性。尿中醛固酮排量增大,但尿17酮及17羟类固醇及ACTH试验正常。
其他辅助检查
常规X线及B超检查。
诊断
根据上述临床特征一般诊断不难。诊断要点为: 1.发病年龄早,多在新生儿期。 2.有典型的临床表现,反复呕吐、腹泻,渴感减退或消失,多尿、低渗或等渗性脱水、严重电解质紊乱,部分病人可有酸中毒;患儿生长发育智力发育均落后。 3.使用外源性醋酸脱氧皮质酮或9α-氟氢可的松无反应。 4.实验室检查符合本病条件。即可诊断本病。
治疗
本病临床上以补充食盐为主要治疗手段,部分患儿需用碳酸氢钠纠正酸中毒。治疗有效的指标为病儿失盐状态纠正,渴感恢复,生长发育恢复正常。绝大多数患儿可在2周左右停止治疗。通过补充氯化钠(3~6g/d)可纠正高血钾,降低血浆肾素及醛固酮水平,使临床症状改善。
预后
如早发现、早治疗,预后一般良好。
预防
本病因系遗传性疾病,对本病发生目前尚无有效措施,但对已患病者应积极对症治疗,预防病情发展及并发症发生。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号