概述
膜增生性肾小球肾炎(membrano-proliferative glomerulonephritis,MPGN),是肾小球肾炎中最少见的类型之一,一般分为原发性和继发性。此病曾有多种名字,包括系膜毛细血管性肾小球肾炎(MCGN)、系膜毛细血管增生性肾炎、小叶性肾炎、低补体血症性肾炎等。 本病是一种具有特定病理形态及免疫学表现的综合征。临床主要表现为肾炎、肾病或肾炎肾病同时存在和低补体血症;组织学上可见系膜增生,毛细血管壁增厚,肾小球呈分叶状,故又称分叶性肾炎。其病理改变的主要特点是系膜细胞增生、毛细血管壁增厚及基底膜双轨。根据电镜下电子致密物沉积的部位可将MPGN分为3型:Ⅰ型为内...[详细]
病因
膜增生性肾炎按其临床和实验室特点分为原发性和继发性肾小球病,见表1。 原发性膜增生性肾炎病因不明,一般认为Ⅰ型为免疫复合物病;Ⅱ型为免疫复合物及自身抗体性疾病,可能与遗传有关。 继发性膜增生性肾炎中混合性冷球蛋白血症有3种亚型。Ⅰ型冷球蛋白血症是单株峰球蛋白,通常为骨髓瘤蛋白。Ⅱ型通常为单株峰IgM球蛋白结合IgG,又称抗IgG类风因子,而Ⅲ型则是多株峰免疫球蛋白。Ⅱ型和Ⅲ型冷球蛋白血症易发生肾损害。其病理特征为系膜细胞大量增生、白细胞尤其单核细胞浸润、肾小球基膜增厚有双轨现象。约1/3病例有中小动脉炎,毛细血管内有微血栓形成。MPGN的病因与发病机制不十分明确。Ⅰ型M...[详细]
 原发性膜增生性肾炎病因不明,一般认为Ⅰ型为免疫复合物病;Ⅱ型为免疫复合物及自身抗体性疾病,可能与遗传有关。 继发性膜增生性肾炎中混合性冷球蛋白血症有3种亚型。Ⅰ型冷球蛋白血症是单株峰球蛋白,通常为骨髓瘤蛋白。Ⅱ型通常为单株峰IgM球蛋白结合IgG,又称抗IgG类风因子,而Ⅲ型则是多株峰免疫球蛋白。Ⅱ型和Ⅲ型冷球蛋白血症易发生肾损害。其病理特征为系膜细胞大量增生、白细胞尤其单核细胞浸润、肾小球基膜增厚有双轨现象。约1/3病例有中小动脉炎,毛细血管内有微血栓形成。MPGN的病因与发病机制不十分明确。Ⅰ型MPGN认为是免疫复合物病,由相对大的难溶的免疫复合物反复持续沉积引起。Ⅱ型MPGN患者血清中也存在免疫复合物,冷球蛋白、补体异常、血清C3持续降低。均提示免疫复合物在Ⅱ型MPGN中的作用。Ⅱ型MPGN患者血清中可检出C3肾炎因子(C3NeF),C3NeF是C3bBb转化酶的自身抗体,使C3bBb作用加强,导致补体旁路持续激活,产生持续低补体血症和基膜变性。所以补体代谢障碍为中心环节。 另外,Ⅱ型MPGN肾移植中常复发,可能因病人血清中存在能引起异常糖蛋白形成的物质沉积于基底膜而导致肾炎。 本病可能与遗传有关,Ⅱ型MPGN患者常出现HLA-B7。大多Ⅰ型MPGN病人具有特殊B细胞同种抗原。
原发性膜增生性肾炎病因不明,一般认为Ⅰ型为免疫复合物病;Ⅱ型为免疫复合物及自身抗体性疾病,可能与遗传有关。 继发性膜增生性肾炎中混合性冷球蛋白血症有3种亚型。Ⅰ型冷球蛋白血症是单株峰球蛋白,通常为骨髓瘤蛋白。Ⅱ型通常为单株峰IgM球蛋白结合IgG,又称抗IgG类风因子,而Ⅲ型则是多株峰免疫球蛋白。Ⅱ型和Ⅲ型冷球蛋白血症易发生肾损害。其病理特征为系膜细胞大量增生、白细胞尤其单核细胞浸润、肾小球基膜增厚有双轨现象。约1/3病例有中小动脉炎,毛细血管内有微血栓形成。MPGN的病因与发病机制不十分明确。Ⅰ型MPGN认为是免疫复合物病,由相对大的难溶的免疫复合物反复持续沉积引起。Ⅱ型MPGN患者血清中也存在免疫复合物,冷球蛋白、补体异常、血清C3持续降低。均提示免疫复合物在Ⅱ型MPGN中的作用。Ⅱ型MPGN患者血清中可检出C3肾炎因子(C3NeF),C3NeF是C3bBb转化酶的自身抗体,使C3bBb作用加强,导致补体旁路持续激活,产生持续低补体血症和基膜变性。所以补体代谢障碍为中心环节。 另外,Ⅱ型MPGN肾移植中常复发,可能因病人血清中存在能引起异常糖蛋白形成的物质沉积于基底膜而导致肾炎。 本病可能与遗传有关,Ⅱ型MPGN患者常出现HLA-B7。大多Ⅰ型MPGN病人具有特殊B细胞同种抗原。发病机制
MPGN的发病机制尚不清楚,目前认为与免疫学机制有关。50%~60%的MPGN患者血中出现补体C3、C1q及C4降低,提示旁路途径及经典途径均被激活而导致血中补体的降低。并伴有免疫复合物的轻度增多及冷球蛋白血症,肾小球内有免疫球蛋白及补体的沉积。但补体的异常与疾病的关系、免疫复合物的作用还有待进一步探讨。 根据各种免疫复合物沉积在肾小球基底膜内和系膜区的形式及沉积的程度不同将MPGN分为3种类型。 1.Ⅰ型 以内皮下及系膜区的复合物沉积为主。Ⅰ型与病毒、细菌及寄生虫感染及一些免疫复合物疾病有关(如遗传性补体缺失、SLE、混合性冷球蛋白血症、SBE、分流性肾炎、淋巴瘤、血吸...[详细]
临床表现
本组疾病在原发性肾小球病中较少见,也是肾病综合征中为数不多的增殖性肾炎之一。各种病理类型的临床表现基本相似,无论本病的临床表现为何种综合征,几乎都有蛋白尿和血尿同时存在,蛋白尿为非选择性,血尿常为镜下持续性血尿,有10%~20%患者常于呼吸道感染后出现发作性肉眼血尿,为严重的、多样尿红细胞畸形的肾小球源性血尿。约1/3以上患者伴有高血压,高血压的程度一般比较轻,但也有个别病例,尤其是Ⅱ型患者,可能发生严重的高血压,大剂量的激素治疗也有可能诱发高血压危象。至少有半数的患者出现急性或慢性肾功能不全,在发病初期出现肾功能不全常提示预后不良。患者常于起病后即有较严重的正细胞正色素性贫血,表现为面色苍白...[详细]
并发症
1.感染 本病表现为肾病综合征时,大量蛋白质丢失、营养不良、免疫功能紊乱及应用糖皮质激素治疗均可使机体抵抗力下降,诱发感染性疾病发生,且临床征象常不明显,虽有多种抗生素供选择,若治疗不及时或不彻底,仍易引起肾病综合征复发和病情加重,甚至导致病人死亡。 2.血栓、栓塞并发症 由于血液浓缩(有效血容量减少)及高脂血症造成血液黏稠度增加;蛋白质大量丢失及肝脏代偿性合成蛋白增加,会引起机体凝血、抗凝和纤溶系统失衡。另外,肾病综合征时血小板功能亢进,加之应用利尿药和糖皮质激素等,均可加重高凝状态,容易发生血栓、栓塞并发症。其中肾静脉血栓最常见,3/4病例因形成缓慢,故临床症状不显。此外,肺...[详细]
实验室检查
本病患者几乎总有血尿,包括镜下或者肉眼血尿。蛋白尿可以比较轻微,约有30%表现为无症状性蛋白尿,但半数患者尿蛋白>3.5g/24h,90%以上患者蛋白尿选择性差。尿FDP和C3可升高。 实验室检查的一个特征性改变就是血补体的降低。约有75%的本病患者C3持续性降低,其中Ⅱ型病变中较常见,占80%~90%,约10%的患者显著下降到低于20~30mg/dl。在Ⅰ型病变中,平均C3浓度降至正常的68%,在Ⅱ型降为正常的47%,而且Ⅱ型比Ⅰ型持续时间更长。早期起作用的补体成分(如C1q、CA),在Ⅰ型病变中有不同程度的下降,而在Ⅱ型中通常正常或者轻度下降,但Ⅱ型常伴有晚期起作用的补体成分C5...[详细]
其他辅助检查
1.Ⅰ型膜增殖性肾小球肾炎病理及活检检查 (1)光镜:Ⅰ型膜增殖性肾小球肾炎主要改变是弥漫性的毛细血管壁增厚和血管内细胞增生,还有单个核白细胞和中性粒细胞浸润。系膜区和毛细血管壁由于细胞增生和基质增加而呈现不同程度的扩张,通常是均一地影响几乎所有的小叶,可引起毛细血管丛的分叶结构突出,因此早期称这一病变为小叶性肾小球肾炎。至于分叶型和非分叶型病变之间是否存在因果或先后关系,至今尚无定论。系膜区明显扩张形成结节状,结节中间区可有硬化灶,与糖尿病肾小球硬化或者轻链沉积病的病变相似,但是结合光镜、免疫荧光和电镜的结果就可以容易地将本病和其他疾病区分开来。另一明显但不是特异的表现是肾小球基底...[详细]
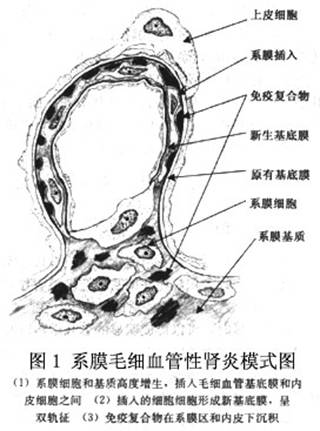 (2)电镜:超微结构的典型特征是系膜细胞和基质在肾小球毛细血管基膜与内皮细胞之间的伸展和间位,有电子致密免疫复合物沉积。系膜毛细血管性肾小球肾炎这一名称正是由Ⅰ型病变中系膜和毛细血管的这种改变而来。在内皮下沉积物的周围和系膜细胞胞浆附近区域,可见有新的基底膜物质形成。在系膜细胞增生和系膜基质扩展的区域通常有散在的致密物沉积。上皮下可有多少不一的电子致密物沉积,当数量足够多时与膜性肾病相似,一些肾脏病理学家称之为“混合性膜性和增生性肾小球肾炎”,或者Burkholder提出的“Ⅲ型系膜毛细血管性肾小球肾炎”。有极少数病变其肾小球损害与Ⅰ型的光镜和免疫荧光相似,但超微结构以肾小球基底膜不规则增厚,膜内有密度不一的大量沉积物为特征,这类病变也归入Ⅲ型。系膜基质和基底膜之间可有单核细胞或中性粒细胞浸润。有些肾活检组织中有少至中量的膜外沉积物呈“驼峰”状。上皮足突常消失。光镜下的透明血栓表现为血管腔内球性致密物,当这些结构或者任何其他电子致密沉积物呈现微管样结构,提示可能为冷球蛋白血症或者免疫触须样肾病。 (3)免疫荧光:特征性的改变是补体尤其是C3和免疫球蛋白呈颗粒状或带状分布,可显示出小叶外周的轮廓,这与电镜观察到的内皮下免疫复合物沉积的部位相一致,沉积物的形态通常不如膜性肾病对称,颗粒状也没有那么明显。备解素及B因子呈相似分布。系膜的颗粒状沉积可以明显也可以不明显。少数Ⅰ型可见免疫复合物沿小管基底膜和(或)肾小球外的血管沉着。沉积的免疫复合物的成分可有很大不同,可能反映了引起Ⅰ型的多种原因,大多数患者C3的沉积比任何免疫球蛋白都明显;有一些以IgG或IgM为主;还有极少数以IgA为主,可以认为是表现为系膜毛细血管性肾小球肾炎的IgA肾病。早期起作用的补体成分如C1q和C4,比C3稍少见。少数病人可见毛细血管壁有Ig(尤其是IgM和IgG)呈节段性颗粒状分布,偶尔也可见于系膜区。毛细血管腔内大量的免疫球蛋白和补体沉积形成球状结构,这与光镜观察的透明血栓相一致,提示病变继发于系统性红斑狼疮或者冷球蛋白血症。 2.Ⅱ型膜增生性肾小球肾炎病理及活检检查 (1)光镜:Ⅱ型的光镜改变比Ⅰ型变化更多,不只是膜增殖的改变,这使一些肾脏病理学家认为称之为致密物沉积病比Ⅱ型系膜毛细血管性肾小球肾炎更准确。1995年WH0将其分类为继发性代谢性疾病中。在组织学上,表现为肾小球系膜细胞和基质增生,增生明显时,可以形成明显的分叶结构以及毛细血管壁增厚。有些毛细血管因系膜间位,使毛细血管壁呈双轨状。这些典型的膜增殖性改变与Ⅰ型相似,但是部分有明显的毛细血管壁增厚,细胞增生呈灶状或者不伴有细胞增生,还有部分仅有细胞呈灶状或者弥漫增生,但没有毛细血管壁的明显增厚。系膜改变的程度有很大的个体差异性,系膜细胞和基质的增加可以很轻微也可以很严重。用Masson三色染色在系膜区常可见有圆形嗜伊红沉积物,有些可有上皮下“驼峰”状沉积物。毛细血管腔内中性白细胞数常增加,少数有新月体形成,间质可有白细胞浸润和纤维化。因此,Ⅱ型的光镜改变可以与其他肾炎类似,需要结合电镜和免疫荧光的结果才能准确判断。有个别报告此型患者不伴有系膜增生性改变,因而与Ⅰ型不同。 (2)电镜:Ⅱ型又叫做致密物沉积病,强调对本病具有诊断性的特征是肾小球基底膜上不连续的电子致密带形成,并伴有系膜球状或不规则状致密物沉积,有时内皮下和上皮下也有沉积,一些改变与链球菌感染后肾小球肾炎的“驼峰”样相似。基底膜明显增宽和有极度电子致密结构,这有很大的诊断意义。但在每一个肾小球中,有些毛细血管壁可没有上述病损。致密结构可呈梭状、球状或香肠状,与正常结构之间的分界很清晰。系膜细胞和基质常向外周伸展和间位,但不及Ⅰ型明显。上皮细胞足突常完全消失。许多病人系膜区常有圆形的电子致密沉积物。如肾小管基底膜有电子致密沉积物,则高度提示为Ⅱ型病损。 (3)免疫荧光:大量C3在肾小球毛细血管壁基底膜呈线状或带状沉积,C3呈不连续的线状类型,可以显示出毛细血管壁、肾小球囊和肾小管的轮廓。系膜的沉积物呈分散的针状或环状,环状是由于仅沉积物的外侧被染色的结果。另外,许多毛细血管壁可有颗粒状C3沉积物,线状毛细血管壁荧光呈双轨状,是由于C3沉积于基底膜的两侧。其他补体成分仅见于不到50%的活检病例。免疫球蛋白沉积很少。 3.其他类型系膜毛细血管性肾小球肾炎 目前尚不能确定它们是Ⅰ型病损的变异类型,还是独立的病变。这些类型几乎都以电镜观察为基础加以识别。Burkholder提出了Ⅲ型病损,它的特征是除了与Ⅰ型的共同病理改变之外,尚有较突出的上皮下免疫复合物沉着,并有细血管壁伴有孤立的膜外沉积物,被基膜物质突起所隔离(类似膜性肾小球肾炎基底膜的钉状突起),有些学者认为此类型是膜性和增生性肾小球肾炎的混合型(表2)。此外,近年来还有一些学者报道了各种各样的一些变异类型,如Ⅳ型,以基底膜分裂呈层状为特点,伴有上皮下和内皮下沉积物。其余在此不予赘述。
(2)电镜:超微结构的典型特征是系膜细胞和基质在肾小球毛细血管基膜与内皮细胞之间的伸展和间位,有电子致密免疫复合物沉积。系膜毛细血管性肾小球肾炎这一名称正是由Ⅰ型病变中系膜和毛细血管的这种改变而来。在内皮下沉积物的周围和系膜细胞胞浆附近区域,可见有新的基底膜物质形成。在系膜细胞增生和系膜基质扩展的区域通常有散在的致密物沉积。上皮下可有多少不一的电子致密物沉积,当数量足够多时与膜性肾病相似,一些肾脏病理学家称之为“混合性膜性和增生性肾小球肾炎”,或者Burkholder提出的“Ⅲ型系膜毛细血管性肾小球肾炎”。有极少数病变其肾小球损害与Ⅰ型的光镜和免疫荧光相似,但超微结构以肾小球基底膜不规则增厚,膜内有密度不一的大量沉积物为特征,这类病变也归入Ⅲ型。系膜基质和基底膜之间可有单核细胞或中性粒细胞浸润。有些肾活检组织中有少至中量的膜外沉积物呈“驼峰”状。上皮足突常消失。光镜下的透明血栓表现为血管腔内球性致密物,当这些结构或者任何其他电子致密沉积物呈现微管样结构,提示可能为冷球蛋白血症或者免疫触须样肾病。 (3)免疫荧光:特征性的改变是补体尤其是C3和免疫球蛋白呈颗粒状或带状分布,可显示出小叶外周的轮廓,这与电镜观察到的内皮下免疫复合物沉积的部位相一致,沉积物的形态通常不如膜性肾病对称,颗粒状也没有那么明显。备解素及B因子呈相似分布。系膜的颗粒状沉积可以明显也可以不明显。少数Ⅰ型可见免疫复合物沿小管基底膜和(或)肾小球外的血管沉着。沉积的免疫复合物的成分可有很大不同,可能反映了引起Ⅰ型的多种原因,大多数患者C3的沉积比任何免疫球蛋白都明显;有一些以IgG或IgM为主;还有极少数以IgA为主,可以认为是表现为系膜毛细血管性肾小球肾炎的IgA肾病。早期起作用的补体成分如C1q和C4,比C3稍少见。少数病人可见毛细血管壁有Ig(尤其是IgM和IgG)呈节段性颗粒状分布,偶尔也可见于系膜区。毛细血管腔内大量的免疫球蛋白和补体沉积形成球状结构,这与光镜观察的透明血栓相一致,提示病变继发于系统性红斑狼疮或者冷球蛋白血症。 2.Ⅱ型膜增生性肾小球肾炎病理及活检检查 (1)光镜:Ⅱ型的光镜改变比Ⅰ型变化更多,不只是膜增殖的改变,这使一些肾脏病理学家认为称之为致密物沉积病比Ⅱ型系膜毛细血管性肾小球肾炎更准确。1995年WH0将其分类为继发性代谢性疾病中。在组织学上,表现为肾小球系膜细胞和基质增生,增生明显时,可以形成明显的分叶结构以及毛细血管壁增厚。有些毛细血管因系膜间位,使毛细血管壁呈双轨状。这些典型的膜增殖性改变与Ⅰ型相似,但是部分有明显的毛细血管壁增厚,细胞增生呈灶状或者不伴有细胞增生,还有部分仅有细胞呈灶状或者弥漫增生,但没有毛细血管壁的明显增厚。系膜改变的程度有很大的个体差异性,系膜细胞和基质的增加可以很轻微也可以很严重。用Masson三色染色在系膜区常可见有圆形嗜伊红沉积物,有些可有上皮下“驼峰”状沉积物。毛细血管腔内中性白细胞数常增加,少数有新月体形成,间质可有白细胞浸润和纤维化。因此,Ⅱ型的光镜改变可以与其他肾炎类似,需要结合电镜和免疫荧光的结果才能准确判断。有个别报告此型患者不伴有系膜增生性改变,因而与Ⅰ型不同。 (2)电镜:Ⅱ型又叫做致密物沉积病,强调对本病具有诊断性的特征是肾小球基底膜上不连续的电子致密带形成,并伴有系膜球状或不规则状致密物沉积,有时内皮下和上皮下也有沉积,一些改变与链球菌感染后肾小球肾炎的“驼峰”样相似。基底膜明显增宽和有极度电子致密结构,这有很大的诊断意义。但在每一个肾小球中,有些毛细血管壁可没有上述病损。致密结构可呈梭状、球状或香肠状,与正常结构之间的分界很清晰。系膜细胞和基质常向外周伸展和间位,但不及Ⅰ型明显。上皮细胞足突常完全消失。许多病人系膜区常有圆形的电子致密沉积物。如肾小管基底膜有电子致密沉积物,则高度提示为Ⅱ型病损。 (3)免疫荧光:大量C3在肾小球毛细血管壁基底膜呈线状或带状沉积,C3呈不连续的线状类型,可以显示出毛细血管壁、肾小球囊和肾小管的轮廓。系膜的沉积物呈分散的针状或环状,环状是由于仅沉积物的外侧被染色的结果。另外,许多毛细血管壁可有颗粒状C3沉积物,线状毛细血管壁荧光呈双轨状,是由于C3沉积于基底膜的两侧。其他补体成分仅见于不到50%的活检病例。免疫球蛋白沉积很少。 3.其他类型系膜毛细血管性肾小球肾炎 目前尚不能确定它们是Ⅰ型病损的变异类型,还是独立的病变。这些类型几乎都以电镜观察为基础加以识别。Burkholder提出了Ⅲ型病损,它的特征是除了与Ⅰ型的共同病理改变之外,尚有较突出的上皮下免疫复合物沉着,并有细血管壁伴有孤立的膜外沉积物,被基膜物质突起所隔离(类似膜性肾小球肾炎基底膜的钉状突起),有些学者认为此类型是膜性和增生性肾小球肾炎的混合型(表2)。此外,近年来还有一些学者报道了各种各样的一些变异类型,如Ⅳ型,以基底膜分裂呈层状为特点,伴有上皮下和内皮下沉积物。其余在此不予赘述。
诊断
本病诊断的主要依据是病理检查结果,电镜和免疫荧光检查可以区分Ⅰ型和Ⅱ型。持续性的低补体血症,持续无选择性的蛋白尿(或肾病综合征)伴有严重的多样畸形的红细胞尿,与肾功能下降不成比例的贫血,常提示该病发生。C3肾炎因子和血补体C3同时降低常提示病情活动。低C3血症还见于其他继发性肾小球病如:活动性肝病、白血病、转移性肿瘤、和免疫球蛋白血症,因其有原发病的特征不难鉴别。 由于MPGN常在上呼吸道感染之后急性起病,表现为急性肾炎综合征,甚至半数左右患者抗“O”链球菌感染的证据呈阳性,故应与链球菌感染后肾小球肾炎相鉴别。后者常有肉眼血尿,而血补体水平在2个月内常恢复正常。本病肉眼血尿在发病后第...[详细]
治疗
本病所致肾病综合征的治疗常常比较困难。小剂量、隔天泼尼松治疗可能有利于改善肾功能。West等使用隔天口服激素长期治疗,在治疗前后比较肾活检,结果证明此法有利于肾脏的存活。目前大部分肾病学家仅做对症治疗。 Ⅰ型的治疗,除糖皮质激素外,还可用其他药物如免疫抑制药和抗凝剂。 对于各年龄段MPGN患者,如肾功能正常且仅表现为无症状轻度蛋白尿时,无须接受激素、免疫抑制药治疗。仅需每3~4个月随访1次,密切观察肾功能、蛋白尿和血压控制情况。成人和儿童原发性MPGN患者,在尿蛋白>3g/d,肾功能损害及活检发现肾间质病变时,方可给予激素、免疫抑制药治疗。 对于有蛋白尿(>3g/...[详细]
预后
大量研究证实原发性MPGN 10年肾存活率达60%~65%,而且各型MPGN病程及预后类似。肾病综合征(大量蛋白尿)和出现肾间质病变是预后不良的主要征兆。临床发现表现为持续性高血压、GFR下降的肾病综合征预后差。发病年龄小和小儿患者预后良好;成年人发病者病变进行性加重,预后不良。肾小球系膜细胞增生、基底膜增厚与预后无明显关系,而局灶性新月体形成的多少与预后明显相关,间质改变的轻重程度与预后明显相关,新月体形成及严重的肾小管间质病变预后差。约50%表现为肾病综合征的病人10年内发展为ESRF,50%病人肾病综合征持续数年后消失,肾功能正常。肾移植后,本病可再发,但小于10%病人导致移植肾丧失。接...[详细]
预防
本病3型病程转归基本相同。预防要从自身健康着手,平时避免劳累,合理饮食,科学锻炼,增强体质,提高机体免疫力,以防疾病发生。对于已患和出现并发症的病人,应对原发病及并发症进行积极有效的预防和治疗。一旦发现感染,应及时选用对致病菌敏感、强效且无肾毒性的抗生素治疗,有明确感染灶者应尽快去除,以防肾功能不全的进行性发展。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号