概述
垂体腺瘤是常见的良性颅内内分泌肿瘤。Marie于1886年首先描述肢端肥大症,1887年Minkowski论及肢端肥大症由垂体腺排列异常引起,1900年Benda认识到伴肢端肥大症的嗜酸性腺瘤并证明肿瘤是来自腺垂体细胞的真性肿瘤。1901年Frankel等研究肢端肥大症后提出该症有垂体嫌色细胞的增生以及垂体功能的亢进,而1908年Marburg认识到无分泌垂体腺瘤有垂体功能低下的临床表现。1909年Cushing进一步阐明和解释了分泌性嗜酸性腺瘤引起垂体功能亢进(肢端肥大症)及无功能腺瘤(嫌色细胞瘤)导致垂体功能低下之间的临床关系,并于1912年明确提出垂体高分泌与低分泌的相反症群,描述了它们...[详细]
病因
目前认为垂体腺瘤来源于腺垂体细胞,如单激素细胞腺瘤如生长激素、泌乳素细胞腺瘤等来源于分泌相应激素的腺细胞,而对一些多激素来源的腺瘤还有争议。过去一直认为一种细胞只能分泌一种相应的激素,20世纪70年代Zimmemrman用PAP法研究5例正常人垂体组织证实,在同一种细胞内具有能与生长激素和泌乳素两种激素抗体结合的颗粒,说明两种激素可以同时在同一垂体细胞内产生。Midyley认为促卵泡素和黄体生成素可由同一种细胞分泌。Horvath报告了9例垂体泌乳素、生长激素细胞腺瘤。Kovacs也指出在非肿瘤情况下这种双激素分泌细胞同样散在于腺垂体中,只是数量较少而已。上述研究结果说明,垂体内一种细胞不是只...[详细]
发病机制
垂体腺瘤的发病机制研究可归为两大学说:一为垂体内在异常学说,另一为下丘脑调节机制失常学说。采用分子生物学方法研究后,使长期以来一直有争议的垂体学说和下丘脑学说趋向于统一。目前认为垂体腺瘤的发展可分为两个阶段——起始阶段和促进阶段,即垂体细胞先发生突变,然后在内外因素的促进下突变的细胞增生,发展为垂体腺瘤。 1.垂体内在异常 (1)基因突变:垂体细胞的基因突变在垂体腺瘤发生中的作用近年受到高度重视,目前,已肯定的主要为一些与细胞信号转导有关的基因的点突变,这些基因包括G蛋白Gs的α亚单位(Gsα)基因、ras原癌基因、蛋白激酶C的α亚型(PKCα)基因等。 Gsα...[详细]
临床表现
垂体腺瘤瘤细胞的倍增时间为100~700天,故肿瘤生长缓慢,这种生物学特征决定了垂体腺瘤一般起病潜隐,早期可无症状。有的肿瘤甚至自始至终没有症状,死后尸检始被发现。垂体腺瘤主要有颅内神经功能障碍及内分泌功能障碍两方面表现: 1.神经功能障碍 垂体腺瘤引起的神经症状直接与肿瘤大小及其生长方向有关。一般无分泌功能腺瘤在确诊时往往肿瘤体积已较大,多向鞍上及鞍外生长,临床神经症状多较明显。分泌性腺瘤因早期产生内分泌亢进症状,确诊时大多体积较小,肿瘤多位于蝶鞍内或轻微向鞍上生长,临床不产生或仅有轻微的神经症状。 (1)头痛:约2/3无分泌性垂体腺瘤病人可有头痛,但不太严重。早期头痛...[详细]
并发症
1.手术治疗的并发症 (1)鞍内并发症:包括颈内动脉损伤(占0.4%~1.4%),可引起假性动脉瘤、颈内动脉-海绵窦瘘,术后大血管痉挛、闭塞,及脑神经损伤(占0.4%~1.9%),尤以展神经损伤为多见。 (2)鞍上操作所致并发症:包括下丘脑、垂体柄、垂体损伤;视神经、视交叉及周围血管的损伤导致视力减退或失明(占0.4%~2.4%),后者也可由残余肿瘤出血、肿胀、鞍内填塞物过多等原因引起;鞍膈及蛛网膜损伤破裂发生脑脊液漏(占1.5%~4.2%,甚高达9%~15%),可引起气颅、脑膜炎(占0%~2%);其他尚有蛛网膜下腔出血、双额硬膜外血肿、癫痫等。 (3)经蝶入路及...[详细]
实验室检查
垂体腺瘤的常规实验室检查多无异常。对垂体腺瘤来说,重要的是各种内分泌功能检查。由于病变及病程的不同,垂体腺瘤的内分泌功能检查可有不同结果。 1.生长激素(GH) 由垂体GH细胞分泌,受下丘脑调节,疑诊GH腺瘤时,应测GH基础值和葡萄糖抑制试验。禁食12h后,休息情况下的GH正常值2~4μg/L,易受情绪、低血糖、睡眠、体力活动和应激状态等影响,约90%的GH腺瘤病人GH基础值高于10μg/L。GH水平在5~10μg/L可以是GH腺瘤,但个别情况也见于正常人,因此,应做葡萄糖抑制试验。正常人口服葡萄糖100g后2h,GH低于正常值,3~4h后回升。GH腺瘤病人不受此影响,呈不能抑制现...[详细]
其他辅助检查
1.垂体微腺瘤的CT和MRI表现 (1)直接征象:垂体内低密度(信号)区是诊断垂体微腺瘤的可靠征象(图1)。低密度(信号)区在3mm以上或超过垂体体积的1/3即可诊断为垂体微腺瘤。低密度(信号)区的显示与垂体及肿瘤的造影剂充盈方式有关。造影剂快速增强扫描时,由于垂体的血供极其丰富,且无血-脑脊液屏障,注入造影剂后可立即增强,其增强的程度与海绵窦及颈内动脉相接近。而肿瘤组织的血供不及垂体丰富,增强不及垂体迅速,肿瘤密度(信号)增加缓慢,因而在注入造影剂的一瞬间,肿瘤与邻近垂体组织或海绵窦相比呈低密度(信号)。随着时间的推移,循环血中的造影剂浓度逐渐降低,垂体与海绵窦的密度(信号)均逐渐...[详细]
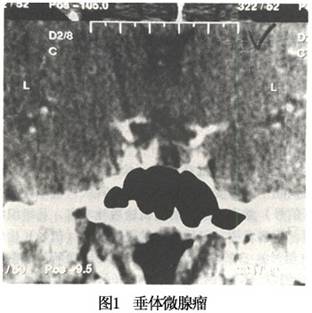 (2)占位征象: ①垂体增高和(或)上缘膨隆:垂体高度超过8mm即提示可能存在微腺瘤。但正常垂体高度也可能>8mm。另外,垂体高度正常也不能否定微腺瘤的存在,因此不能单纯用垂体高度作为微腺瘤是否存在的惟一标准,必须结合其他CT表现。垂体增高且上缘膨隆,则高度提示微腺瘤的存在,若垂体上缘的隆起不对称,则更支持微腺瘤的诊断。有人报道,垂体增高且上缘隆起不对称,91%有肿瘤存在。垂体上缘呈普遍性隆起只有部分病例中线区有肿瘤存在。因为正常垂体上缘也可膨隆,故观察垂体上缘形态也需结合其他征象。 ②垂体柄移位:肿瘤的占位效应可将垂体柄推向对侧,但在少数情况下,垂体柄也可向肿瘤同侧移位。另外,动态增强扫描可见垂体柄周围毛细血管丛,微腺瘤的占位效应也可导致此毛细血管丛的移位。垂体柄偏离中线2mm以上,常常提示微腺瘤的存在。同样,在分析垂体柄的变化时也需结合其他CT征象,因为微腺瘤病人垂体柄可以不移位,而正常人的垂体柄又可略偏离中线。 ③神经垂体消失:冠状CT扫描在通过垂体后缘的层面上,在鞍背前方常可见到略低密度的卵圆形后叶;而MRI检查可更清晰地显示神经垂体。微腺瘤的占位效应常导致后叶受压缩小而不能显示,或被挤向一侧。但若肿瘤发生于前叶前部,体积又较小,其占位效应不重,则仍可见到后叶。故神经垂体消失常常提示有微腺瘤,而后叶显示良好也不能完全排除微腺瘤。 ④鞍底骨质的变化:微腺瘤可导致鞍底骨质的吸收或破坏,使鞍底两侧厚度不一,CT表现为鞍底一侧变薄或破坏。但正常人鞍底厚度有较大变异,只有骨质改变伴有相应部位的其他异常表现时,才可认为异常。 总之,垂体是否异常或是否存在微腺瘤,应从垂体高度、上缘形态、内部密度(信号)、异常密度(信号)区的存在及其大小、密度(信号)及边界、垂体柄的移位、神经垂体及鞍底骨质的变化等几方面进行仔细观察,垂体柄移位、后叶消失及鞍底骨质的变化,可提示有微腺瘤存在。 2.垂体大腺瘤的CT和MRI表现 CT和MRI检查是诊断垂体腺瘤最主要的影像学方法,不仅可以做出定性诊断,而且还可以了解肿瘤的大小、形态、质地以及与周围结构之间的关系(图2),为治疗方法的选择提供和依据。
(2)占位征象: ①垂体增高和(或)上缘膨隆:垂体高度超过8mm即提示可能存在微腺瘤。但正常垂体高度也可能>8mm。另外,垂体高度正常也不能否定微腺瘤的存在,因此不能单纯用垂体高度作为微腺瘤是否存在的惟一标准,必须结合其他CT表现。垂体增高且上缘膨隆,则高度提示微腺瘤的存在,若垂体上缘的隆起不对称,则更支持微腺瘤的诊断。有人报道,垂体增高且上缘隆起不对称,91%有肿瘤存在。垂体上缘呈普遍性隆起只有部分病例中线区有肿瘤存在。因为正常垂体上缘也可膨隆,故观察垂体上缘形态也需结合其他征象。 ②垂体柄移位:肿瘤的占位效应可将垂体柄推向对侧,但在少数情况下,垂体柄也可向肿瘤同侧移位。另外,动态增强扫描可见垂体柄周围毛细血管丛,微腺瘤的占位效应也可导致此毛细血管丛的移位。垂体柄偏离中线2mm以上,常常提示微腺瘤的存在。同样,在分析垂体柄的变化时也需结合其他CT征象,因为微腺瘤病人垂体柄可以不移位,而正常人的垂体柄又可略偏离中线。 ③神经垂体消失:冠状CT扫描在通过垂体后缘的层面上,在鞍背前方常可见到略低密度的卵圆形后叶;而MRI检查可更清晰地显示神经垂体。微腺瘤的占位效应常导致后叶受压缩小而不能显示,或被挤向一侧。但若肿瘤发生于前叶前部,体积又较小,其占位效应不重,则仍可见到后叶。故神经垂体消失常常提示有微腺瘤,而后叶显示良好也不能完全排除微腺瘤。 ④鞍底骨质的变化:微腺瘤可导致鞍底骨质的吸收或破坏,使鞍底两侧厚度不一,CT表现为鞍底一侧变薄或破坏。但正常人鞍底厚度有较大变异,只有骨质改变伴有相应部位的其他异常表现时,才可认为异常。 总之,垂体是否异常或是否存在微腺瘤,应从垂体高度、上缘形态、内部密度(信号)、异常密度(信号)区的存在及其大小、密度(信号)及边界、垂体柄的移位、神经垂体及鞍底骨质的变化等几方面进行仔细观察,垂体柄移位、后叶消失及鞍底骨质的变化,可提示有微腺瘤存在。 2.垂体大腺瘤的CT和MRI表现 CT和MRI检查是诊断垂体腺瘤最主要的影像学方法,不仅可以做出定性诊断,而且还可以了解肿瘤的大小、形态、质地以及与周围结构之间的关系(图2),为治疗方法的选择提供和依据。 非增强扫描可见蝶鞍扩大,鞍底和鞍背骨质吸收变薄、倾斜;肿瘤位于脑外,由鞍内向鞍上生长,占据鞍上池、第三脑室前部甚至达室间孔水平,但极少因此出现梗阻性脑积水;肿瘤可呈实体性或囊实性,无钙化,边界清楚,呈类圆形或哑铃型;两侧海绵窦受肿瘤推移挤压外移,少数肿瘤侵袭海绵窦,包绕颈内动脉甚至使该侧海绵窦明显外移;有时肿瘤可明显向额叶或颞叶发展,或者突入蝶窦。增强扫描可见实体性肿瘤呈均一中度强化,囊性肿瘤呈周边强化,中小体积肿瘤在肿瘤周边可见残存垂体。 3.普通X线检查:头颅正侧位片可显示蝶鞍形态,但不能显示垂体,因此如果垂体腺瘤仅在鞍内生长而未影响蝶鞍形态,则头颅正侧位片可无异常。如肿瘤侵及蝶鞍则可在头颅正侧位片上形成一系列表现,如蝶鞍扩大;鞍壁脱钙、变薄;前、后床突变细甚至缺如;鞍底变阔、下陷;如肿瘤偏于一侧则可使另一侧鞍底下陷明显,侧位片上呈现出双鞍底。 分层摄影、气脑造影、脑室造影和血管造影对垂体腺瘤的诊断也有一定意义,但由于这些检查多较复杂且有一定的危险性,加之CT和MRI的普及,现已很少使用。 4.PET检查 PET作为一种功能显像技术,自20世纪80年代应用于临床以来已取得很大的成功。PET可提供有关肿瘤生化特征、代谢特性、受体分布及酶学特点等方面的信息,在肿瘤的诊断、治疗等方面均有重要的意义。 PRL瘤及某些无功能腺瘤常有代谢增强,因此,用11C标记的左旋蛋氨酸和18F标记的氟脱氧葡萄糖(18F-fluorodeoxyglucose,18F-FDG)可使其显像。用多巴胺受体激动药治疗后,瘤细胞的代谢降低,其摄取11C-左旋蛋氨酸和18F-FDG的能力也下降。11C标记的多巴胺D2受体拮抗药甲基螺哌隆(methylspiperone)和雷氯必利(raclopride)可使PRL瘤显像,且可预测多巴胺受体激动药的疗效。一般来说,11C标记的甲基螺哌隆和雷氯必利显像者对多巴胺受体激动药的反应良好。
非增强扫描可见蝶鞍扩大,鞍底和鞍背骨质吸收变薄、倾斜;肿瘤位于脑外,由鞍内向鞍上生长,占据鞍上池、第三脑室前部甚至达室间孔水平,但极少因此出现梗阻性脑积水;肿瘤可呈实体性或囊实性,无钙化,边界清楚,呈类圆形或哑铃型;两侧海绵窦受肿瘤推移挤压外移,少数肿瘤侵袭海绵窦,包绕颈内动脉甚至使该侧海绵窦明显外移;有时肿瘤可明显向额叶或颞叶发展,或者突入蝶窦。增强扫描可见实体性肿瘤呈均一中度强化,囊性肿瘤呈周边强化,中小体积肿瘤在肿瘤周边可见残存垂体。 3.普通X线检查:头颅正侧位片可显示蝶鞍形态,但不能显示垂体,因此如果垂体腺瘤仅在鞍内生长而未影响蝶鞍形态,则头颅正侧位片可无异常。如肿瘤侵及蝶鞍则可在头颅正侧位片上形成一系列表现,如蝶鞍扩大;鞍壁脱钙、变薄;前、后床突变细甚至缺如;鞍底变阔、下陷;如肿瘤偏于一侧则可使另一侧鞍底下陷明显,侧位片上呈现出双鞍底。 分层摄影、气脑造影、脑室造影和血管造影对垂体腺瘤的诊断也有一定意义,但由于这些检查多较复杂且有一定的危险性,加之CT和MRI的普及,现已很少使用。 4.PET检查 PET作为一种功能显像技术,自20世纪80年代应用于临床以来已取得很大的成功。PET可提供有关肿瘤生化特征、代谢特性、受体分布及酶学特点等方面的信息,在肿瘤的诊断、治疗等方面均有重要的意义。 PRL瘤及某些无功能腺瘤常有代谢增强,因此,用11C标记的左旋蛋氨酸和18F标记的氟脱氧葡萄糖(18F-fluorodeoxyglucose,18F-FDG)可使其显像。用多巴胺受体激动药治疗后,瘤细胞的代谢降低,其摄取11C-左旋蛋氨酸和18F-FDG的能力也下降。11C标记的多巴胺D2受体拮抗药甲基螺哌隆(methylspiperone)和雷氯必利(raclopride)可使PRL瘤显像,且可预测多巴胺受体激动药的疗效。一般来说,11C标记的甲基螺哌隆和雷氯必利显像者对多巴胺受体激动药的反应良好。诊断
垂体腺瘤的诊断主要依据不同类型腺瘤的临床表现,视功能障碍及其他脑神经和脑损害,以及内分泌检查和放射学检查,典型的病例不难做出垂体腺瘤的分类诊断。但对早期的微腺瘤,临床症状不明显,神经症状轻微,内分泌学检查不典型,又无影像学发现的病例则诊断不易,即使单有临床表现或神经症状或内分泌学或影像学改变或四种均有改变的,亦不一定是垂体腺瘤。所以,既要全面了解病情作多方面的检查,获得资料,综合分析,做出诊断和鉴别诊断,确定是否有肿瘤,是不是垂体腺瘤,还要对肿瘤部位、性质、大小、发展方向和累及垂体周围重要结构的影响程度等进行仔细研究,以便选择治疗方案,制定治疗措施,包括手术入路的选择。
预后
20世纪初经颅垂体腺瘤手术死亡率在10%以上。随着科技的发展,诊疗技术的进步和手术经验的积累,现经颅手术死亡率已下降至4%~5%,有的报道在1.2%~16%。北京协和医院经颅手术死亡率为4.7%,近20多年来开展现代经蝶显微外科技术,手术死亡率又下降至0.4%~2%。据Laws治疗505例中7例死亡,死亡率为1.38%(死于脑膜炎、脑脊液漏、下丘脑损伤、颈内动脉损伤和脑底动脉环闭塞各1例,颅内血肿2例)。Zerves统计国际大宗材料2606例微腺瘤死亡率为0.27%,2677例大腺瘤的死亡率为0.86%。北京协和医院经蝶手术892例,死亡4例,死亡率为0.44%,均为大腺瘤(1例死于复发瘤第2...[详细]
预防
目前无相关资料。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号