-
科室:
神经外科
-
别名:
Recklinghausen病
冯·雷克林豪森病
elehantiasis neuromatosa
-
症状:
胸廓单侧畸形
简单部分发作
-
发病部位:
暂无
-
多发人群:
常染色体显性遗传病家族中常见
-
相关疾病:
暂无
神经纤维瘤病(neurofibromatosis,NF)系发生于神经主干或末梢神经轴索鞘神经膜细胞及神经束膜细胞的良性肿瘤,是一种常染色体显性遗传性疾病。1882年Von Recklinghausen首先报道,故又称Von Recklinghausen病。瘤体多由皮肤神经长出,亦可发生于深部神经、脑神经或内脏神经。多见于皮肤组织,亦可发生在胸、腹腔内。一般在儿时发病,无性别差异,青春期或妊娠期可加速发展,有恶变可能。临床以单发或多发的、无痛性皮下肿物为特征,可伴有智力障碍、癫痫、骨折等系统受侵表现。选择性手术切除是主要的治疗方法。表浅的神经纤维瘤,一般不发生恶变,较深而位于软组织内的神经纤维瘤,有恶变成神经纤维肉瘤的可能。 本病似属中医的“气瘤”范畴。如。医宗金鉴·外科心法〃记载:“软而不坚,皮色正常,随喜怒消长,无寒无热者名气瘤”。又如。外科正宗〃记载:“肺主气,劳伤元气,腠理不密,外寒博而为肿口气瘤”。约50%病例代表新的基因突变,分为;皮肤损伤和内脏损伤。本病临床上可分为以下数型:经典型神经纤维瘤;中枢或听神经瘤病;(混合型);(变异型);节段型(皮节型)神经纤维瘤病;无神经纤维瘤;晚发型神经纤维瘤。[收起]
神经纤维瘤病(neurofibromatosis,NF)系发生于神经主干或末梢神经轴索鞘神经膜细胞及神经束膜细胞的良性肿瘤,是一种常染色体显性遗传性疾病。1882年Von Recklinghausen首先报道,故又称Von Recklinghausen病。瘤体多由皮肤神经长出,亦可发生于深部神经、脑神经或内脏神经。多见于皮肤组织,亦可发生在胸、腹腔内。一般在儿时发病,无性别差异,青春期或妊娠期可加速发展,有恶变可能。临床以单发或多发的、无痛性皮下肿物为特征,可伴有智力障碍、癫痫、骨折等系统受侵表现。选择性手术切除是主要的治疗方法。表浅的神经纤维瘤,一般不发生恶变,较深而位于软组织内的神经纤维瘤...[详细]
NF是基因缺陷使神经嵴细胞发育异常导致多系统损害,可归类于神经皮肤综合征,根据临床表现和基因定位,分为神经纤维瘤病Ⅰ型(NFⅠ)和Ⅱ型(NFⅡ)。NFⅠ主要特征为皮肤牛奶咖啡斑和周围神经多发性神经纤维瘤,外显率高,基因位于染色体17q11.2。NFⅡ又称中枢神经纤维瘤或双侧听神经瘤病,基因位于染色体22q。本病为常染色体显性遗传疾病,患者子女中约半数可发病。NF的发生可能由于基因突变,使具有生长调节的基因功能丧失,从而使该细胞失去控制而增生为肿瘤。是生长在神经干处的以纤维细胞为主的良性肿瘤。NFⅠ基因组跨度350Kb,cDNA长11Kb,含59个外显子,编码2818个氨基酸,组成327kD的神经纤维素蛋白(neutofibronin),分布在神经元。除5型NF可能为后合子体细胞突变(post-zygotic somatic mutation)外,约50%病例代表新的基因突变。NFⅠ基因是一肿瘤抑制基因,发生易位、缺失、重排或点突变时肿瘤抑制功能丧失而致病。1型NF的基因在染色体17q11.2的中心周围区,该基因编码的神经纤维瘤素(neurofibromin)是一种由ras蛋白转变来的蛋白,具有生长调节机能。2型NF的基因位于染色体22q11-q13的长臂上,其基因编码的神经鞘瘤素(schwannomin)是一种将肌动蛋白支架(aclincytoskeleton)连接到细胞表面的糖蛋白,亦起着生长调节的作用。NFⅡ基因缺失突变引起Schwann细胞瘤和脑膜瘤。[收起]
NF是基因缺陷使神经嵴细胞发育异常导致多系统损害,可归类于神经皮肤综合征,根据临床表现和基因定位,分为神经纤维瘤病Ⅰ型(NFⅠ)和Ⅱ型(NFⅡ)。NFⅠ主要特征为皮肤牛奶咖啡斑和周围神经多发性神经纤维瘤,外显率高,基因位于染色体17q11.2。NFⅡ又称中枢神经纤维瘤或双侧听神经瘤病,基因位于染色体22q。本病为常染色体显性遗传疾病,患者子女中约半数可发病。NF的发生可能由于基因突变,使具有生长调节的基因功能丧失,从而使该细胞失去控制而增生为肿瘤。是生长在神经干处的以纤维细胞为主的良性肿瘤。NFⅠ基因组跨度350Kb,cDNA长11Kb,含59个外显子,编码2818个氨基酸,组成327kD的神...[详细]
本病的发病机制未明,可能是神经嵴发育异常。晚近认为与神经生长因子生成过多或活性过高,促使神经纤维异常增殖,导致肿瘤生长有关。主要病理特点是外胚层神经组织发育不良、过度增生和肿瘤形成。NFⅠ神经纤维瘤好发于周围神经远端、脊神经根,尤其马尾;脑神经多见于听神经、视神经和三叉神经。脊髓内肿瘤包括室管膜瘤和星型胶质细胞瘤,颅内肿瘤最常见为脑胶质细胞瘤,肿瘤大小不等,成梭性细胞排列,细胞核似栅栏状。 1.大体所见 神经纤维瘤并不都与大神经干相连系,亦可起于很小的无髓纤维。有疏松透亮薄包膜,其外无或有轻反应区。在侵袭性纤维瘤病可浸润周围正常组织,钝性剥离很难达到囊外边缘囊块切除。如肿瘤连于主要神经干上,常侵及神经组织,在囊外分离时,可见神经纤维进入及穿出肿瘤,而不像神经鞘瘤,不侵及神经纤维,不损伤神经很难整块切除。大肿瘤多为Ⅲ期病变,瘤内退变成囊腔含有黄色液体者亦常见。神经纤维瘤无包膜,但界限较清楚。位于真皮和皮下组织内,瘤体主要由神经膜细胞和神经鞘细胞组成,并可见很多增生的神经轴索和丰富的小血管。其中的纤维组织较细,排列紧密,轻度卷曲而成波浪状(如图1所示)。有时可见纤维发生黏液变性。牛奶咖啡色斑中表皮基底细胞层黑色素增多,可见巨大的色素颗粒。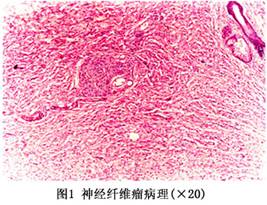 2.镜下形态 电镜检查表明这些肿瘤是由成纤维细胞或周围神经的神经膜细胞增生而形成的。主要表现是疏松的梭形细胞产生细纤维状嗜伊红基质,呈起伏的波浪状,有规律性,有吞噬细胞,含有脂质及含铁血黄素;亦有Verocay小体、血管增生、成熟脂肪、成熟纤维结节等,称为混合型神经纤维瘤。NFⅡ多见双侧听神经瘤和多发性脑膜瘤,瘤细胞排列松散,常见巨核细胞。 组织病理检查可见两类病理(图2)改变。
2.镜下形态 电镜检查表明这些肿瘤是由成纤维细胞或周围神经的神经膜细胞增生而形成的。主要表现是疏松的梭形细胞产生细纤维状嗜伊红基质,呈起伏的波浪状,有规律性,有吞噬细胞,含有脂质及含铁血黄素;亦有Verocay小体、血管增生、成熟脂肪、成熟纤维结节等,称为混合型神经纤维瘤。NFⅡ多见双侧听神经瘤和多发性脑膜瘤,瘤细胞排列松散,常见巨核细胞。 组织病理检查可见两类病理(图2)改变。 (1)皮肤神经纤维瘤:肿瘤无包膜,由神经衣细胞和神经鞘细胞构成,神经衣细胞为未成熟的胶原纤维束,束内原纤维较细,有些纤维间有黏液。神经鞘细胞呈细长菱形或略弯曲呈波形,胞界不清,胞质呈淡嗜伊红性,两端有明显的长短不一的丝状突;胞核常深染,大都与胶原纤维束疏松平行排列呈波形或涡纹状。 (2)皮下丛状神经纤维瘤:侵犯周围大神经,并见不规则形神经束。增生的神经鞘细胞和胶原纤维组成弯曲的条索,周围为黏液样无定形间质。 免疫组化神经纤维瘤根据其主要细胞类型而有不同的抗原表达。S-100蛋白及4型胶原对神经鞘细胞呈阳性表达,表皮膜抗原对神经衣细胞呈阳性表达,vimentin对成纤维细胞和神经鞘细胞呈阳性表达,神经丝和髓磷脂碱性蛋白对轴突和髓磷脂鞘呈阳性表达。 此外,本病还可有脑膜膨出、脊髓空洞症、和先天性畸形等病变。有些病人尚有神经系统以外的病损,如代谢性骨病引起骨质增生、颅孔闭塞、因正常骨质被成纤维细胞和纤维细胞所取代而使骨质稀疏、囊肿形成;以及先天性脊柱异常、骨囊肿、胫骨假关节形成;也可有某一肢体及半侧舌或面部的肥大、脊柱侧弯等。还有报告大脑皮质组织学异常、灰质异位岛区和局限性神经胶质增生等,这可能是产生智力迟钝的原因。 肿瘤通常为良性,且生长缓慢。大约3%~4%可发生恶变,尤其大型丛状神经瘤更有恶变可能。恶变多为周围性肿瘤,中枢的肿瘤极少有恶变。皮肤纤维瘤和纤维软瘤系由纤维组织增生所形成。多位于真皮或皮下组织,无细胞膜,皮肤色素斑由表皮基底细胞层内黑色素沉积所致。[收起]
本病的发病机制未明,可能是神经嵴发育异常。晚近认为与神经生长因子生成过多或活性过高,促使神经纤维异常增殖,导致肿瘤生长有关。主要病理特点是外胚层神经组织发育不良、过度增生和肿瘤形成。NFⅠ神经纤维瘤好发于周围神经远端、脊神经根,尤其马尾;脑神经多见于听神经、视神经和三叉神经。脊髓内肿瘤包括室管膜瘤和星型胶质细胞瘤,颅内肿瘤最常见为脑胶质细胞瘤,肿瘤大小不等,成梭性细胞排列,细胞核似栅栏状。 1.大体所见 神经纤维瘤并不都与大神经干相连系,亦可起于很小的无髓纤维。有疏松透亮薄包膜,其外无或有轻反应区。在侵袭性纤维瘤病可浸润周围正常组织,钝性剥离很难达到囊外边缘囊块切除。如肿瘤连于主要...[详细]
(1)皮肤神经纤维瘤:肿瘤无包膜,由神经衣细胞和神经鞘细胞构成,神经衣细胞为未成熟的胶原纤维束,束内原纤维较细,有些纤维间有黏液。神经鞘细胞呈细长菱形或略弯曲呈波形,胞界不清,胞质呈淡嗜伊红性,两端有明显的长短不一的丝状突;胞核常深染,大都与胶原纤维束疏松平行排列呈波形或涡纹状。 (2)皮下丛状神经纤维瘤:侵犯周围大神经,并见不规则形神经束。增生的神经鞘细胞和胶原纤维组成弯曲的条索,周围为黏液样无定形间质。 免疫组化神经纤维瘤根据其主要细胞类型而有不同的抗原表达。S-100蛋白及4型胶原对神经鞘细胞呈阳性表达,表皮膜抗原对神经衣细胞呈阳性表达,vimentin对成纤维细胞和神经鞘细胞呈阳性表达,神经丝和髓磷脂碱性蛋白对轴突和髓磷脂鞘呈阳性表达。 此外,本病还可有脑膜膨出、脊髓空洞症、和先天性畸形等病变。有些病人尚有神经系统以外的病损,如代谢性骨病引起骨质增生、颅孔闭塞、因正常骨质被成纤维细胞和纤维细胞所取代而使骨质稀疏、囊肿形成;以及先天性脊柱异常、骨囊肿、胫骨假关节形成;也可有某一肢体及半侧舌或面部的肥大、脊柱侧弯等。还有报告大脑皮质组织学异常、灰质异位岛区和局限性神经胶质增生等,这可能是产生智力迟钝的原因。 肿瘤通常为良性,且生长缓慢。大约3%~4%可发生恶变,尤其大型丛状神经瘤更有恶变可能。恶变多为周围性肿瘤,中枢的肿瘤极少有恶变。皮肤纤维瘤和纤维软瘤系由纤维组织增生所形成。多位于真皮或皮下组织,无细胞膜,皮肤色素斑由表皮基底细胞层内黑色素沉积所致。[收起]
本病的发病机制未明,可能是神经嵴发育异常。晚近认为与神经生长因子生成过多或活性过高,促使神经纤维异常增殖,导致肿瘤生长有关。主要病理特点是外胚层神经组织发育不良、过度增生和肿瘤形成。NFⅠ神经纤维瘤好发于周围神经远端、脊神经根,尤其马尾;脑神经多见于听神经、视神经和三叉神经。脊髓内肿瘤包括室管膜瘤和星型胶质细胞瘤,颅内肿瘤最常见为脑胶质细胞瘤,肿瘤大小不等,成梭性细胞排列,细胞核似栅栏状。 1.大体所见 神经纤维瘤并不都与大神经干相连系,亦可起于很小的无髓纤维。有疏松透亮薄包膜,其外无或有轻反应区。在侵袭性纤维瘤病可浸润周围正常组织,钝性剥离很难达到囊外边缘囊块切除。如肿瘤连于主要...[详细]
1.本病临床上可分为以下数型 (1)型NF:为经典型神经纤维瘤,占所有NF患者的85%以上。患者出现多数神经纤维瘤,大小数毫米至数厘米不等,并出现多数广泛分布的咖啡斑,很少或无神经系统损害。约1/4的6岁以下的患儿和几乎所有老年患者出现虹膜Lisch结节。 (2)型NF:又称中枢或听神经瘤病,与1型的区别为出现双侧听神经瘤。 (3)型NF(混合型)和4型NF(变异型):类似2型NF,但出现较多数的神经纤维瘤。 以上4型发生视神经胶质瘤、神经鞘瘤、脑脊膜瘤的危险性较大,且呈常染色体显性遗传。 (4)型NF:又称节段型(皮节型)神经纤维瘤病。通常为非遗传性,考虑由后合子体细胞突变所致。可呈双侧性。 (5)型NF:无神经纤维瘤,仅见咖啡斑。其诊断为咖啡斑必须在两代中发生。 (6)型NF:又称晚发型神经纤维瘤,20岁后才发生神经纤维瘤,是否为遗传性尚不明。 2.本病的临床表现有如下特点 特征性损害主要为神经纤维瘤,其他依次为咖啡斑、腋部雀斑、巨大色素性毛痣(神经痣)、骶部多毛症、颅回状皮肤和巨舌。 (1)皮肤损害: ①皮肤神经纤维瘤:为真皮肿瘤,表面平坦或突起,半球状或有蒂,颜色呈粉红色、肉色、果红色不等,触之柔软如疝状(图3)。用手指轻压可将柔软性肿瘤推向脂肪层,松开手指则弹回,可与脂肪瘤鉴别。 ②皮下丛状神经纤维瘤:少见。为沿周围神经缓慢长出的结节,呈弥漫性肿胀,界限不清。肿瘤可高度增生,发生皱褶,臃肿下垂。触摸时犹如一袋蠕虫。 ③咖啡斑:为本病的标志性损害。通常呈色素均匀一致的淡褐色斑,不规则圆形或卵圆形,直径1.5~5cm,见于出生1岁的婴儿。出现6个以上,直径至少1.5cm(儿童最小直径为0.5cm)的咖啡斑,对本病有诊断价值,通常提示为l型NF。 ④其他皮肤损害:腋窝部雀斑(crowe征)可伸展至颈部,发生于鼠蹊部者可伸展至会阴部。此外皮肤可见青铜色着色和过度色素沉着。亦有伴发黄色肉芽肿者。 (2)内脏损害: ①骨损害:通常为侵蚀性,可引起脊柱前凸、脊柱后凸、假性关节病、脊柱裂、脱位和非外伤性骨折。 ②虹膜Lisch结节:几见于所有成年患者。 ③内分泌损害:可见肢端肥大症、克汀病、黏液水肿、甲状腺功能亢进、嗜铬细胞瘤、性早熟等。 ④肺损害:少数患者可发生弥漫性间质性肺炎。 ⑤恶性肿瘤:皮肤神经纤维瘤偶可发展为神经纤维肉瘤和恶性神经鞘瘤。并有报道伴发wilm肿瘤、横纹肌肿瘤及慢性骨髓性白血病者。 (3)神经系统损害:可发生智力发育迟缓,痴呆、癫痫和多种颅内恶性肿瘤。 (4)中医辨证:中医认为本病多因先天素质缺陷,或劳伤肺气,腠理不密,外邪所搏,气血不和,阻滞经络而发于肌肤。[收起]
1.本病临床上可分为以下数型 (1)型NF:为经典型神经纤维瘤,占所有NF患者的85%以上。患者出现多数神经纤维瘤,大小数毫米至数厘米不等,并出现多数广泛分布的咖啡斑,很少或无神经系统损害。约1/4的6岁以下的患儿和几乎所有老年患者出现虹膜Lisch结节。 (2)型NF:又称中枢或听神经瘤病,与1型的区别为出现双侧听神经瘤。 (3)型NF(混合型)和4型NF(变异型):类似2型NF,但出现较多数的神经纤维瘤。 以上4型发生视神经胶质瘤、神经鞘瘤、脑脊膜瘤的危险性较大,且呈常染色体显性遗传。 (4)型NF:又称节段型(皮节型)神经纤维瘤病。通常为...[详细]
②皮下丛状神经纤维瘤:少见。为沿周围神经缓慢长出的结节,呈弥漫性肿胀,界限不清。肿瘤可高度增生,发生皱褶,臃肿下垂。触摸时犹如一袋蠕虫。 ③咖啡斑:为本病的标志性损害。通常呈色素均匀一致的淡褐色斑,不规则圆形或卵圆形,直径1.5~5cm,见于出生1岁的婴儿。出现6个以上,直径至少1.5cm(儿童最小直径为0.5cm)的咖啡斑,对本病有诊断价值,通常提示为l型NF。 ④其他皮肤损害:腋窝部雀斑(crowe征)可伸展至颈部,发生于鼠蹊部者可伸展至会阴部。此外皮肤可见青铜色着色和过度色素沉着。亦有伴发黄色肉芽肿者。 (2)内脏损害: ①骨损害:通常为侵蚀性,可引起脊柱前凸、脊柱后凸、假性关节病、脊柱裂、脱位和非外伤性骨折。 ②虹膜Lisch结节:几见于所有成年患者。 ③内分泌损害:可见肢端肥大症、克汀病、黏液水肿、甲状腺功能亢进、嗜铬细胞瘤、性早熟等。 ④肺损害:少数患者可发生弥漫性间质性肺炎。 ⑤恶性肿瘤:皮肤神经纤维瘤偶可发展为神经纤维肉瘤和恶性神经鞘瘤。并有报道伴发wilm肿瘤、横纹肌肿瘤及慢性骨髓性白血病者。 (3)神经系统损害:可发生智力发育迟缓,痴呆、癫痫和多种颅内恶性肿瘤。 (4)中医辨证:中医认为本病多因先天素质缺陷,或劳伤肺气,腠理不密,外邪所搏,气血不和,阻滞经络而发于肌肤。[收起]
1.本病临床上可分为以下数型 (1)型NF:为经典型神经纤维瘤,占所有NF患者的85%以上。患者出现多数神经纤维瘤,大小数毫米至数厘米不等,并出现多数广泛分布的咖啡斑,很少或无神经系统损害。约1/4的6岁以下的患儿和几乎所有老年患者出现虹膜Lisch结节。 (2)型NF:又称中枢或听神经瘤病,与1型的区别为出现双侧听神经瘤。 (3)型NF(混合型)和4型NF(变异型):类似2型NF,但出现较多数的神经纤维瘤。 以上4型发生视神经胶质瘤、神经鞘瘤、脑脊膜瘤的危险性较大,且呈常染色体显性遗传。 (4)型NF:又称节段型(皮节型)神经纤维瘤病。通常为...[详细]
随病情发展,出现的症状体征多样,可以是本病表现,也可以看作并发症。 1.骨折或脱位 肿瘤侵及骨骼系统时,可引起骨折、脱位、脊柱畸形,可并发脊柱侧凸,先天性胫骨假关节和先天性锁骨假关节。肿瘤压迫腓总神经时可致足下垂。 2.癫痫 肿瘤侵及中枢系统时,可有癫痫样发作,应注意防止外伤。 3.神经纤维瘤有时可自行破溃出血,也可发生肿瘤内部的大出血,严重时可引起休克。 4.肢体上巨大的肿瘤可经常发生破溃,导致感染化脓,甚至截肢。 5.肺损害 少数患者可发生弥漫性间质性肺炎。 6.恶性肿瘤 皮肤神经纤维瘤偶可发展为神经纤维肉瘤和恶性神经鞘瘤。并有报道伴发wilm肿瘤、横纹肌肿瘤及慢性骨髓性白血病者。[收起]
随病情发展,出现的症状体征多样,可以是本病表现,也可以看作并发症。 1.骨折或脱位 肿瘤侵及骨骼系统时,可引起骨折、脱位、脊柱畸形,可并发脊柱侧凸,先天性胫骨假关节和先天性锁骨假关节。肿瘤压迫腓总神经时可致足下垂。 2.癫痫 肿瘤侵及中枢系统时,可有癫痫样发作,应注意防止外伤。 3.神经纤维瘤有时可自行破溃出血,也可发生肿瘤内部的大出血,严重时可引起休克。 4.肢体上巨大的肿瘤可经常发生破溃,导致感染化脓,甚至截肢。 5.肺损害 少数患者可发生弥漫性间质性肺炎。 6.恶性肿瘤 皮肤神经纤维瘤偶可发展为神经纤维肉瘤和恶性神经鞘瘤...[详细]
1.染色体检查 常染色体17q、22q异常。基因分析可确定NFⅠ和NFⅡ突变类型。 2.免疫组化 神经纤维瘤根据其主要细胞类型而有不同的抗原表达。S-100蛋白及4型胶原对神经鞘细胞呈阳性表达,表皮膜抗原对神经衣细胞呈阳性表达,vimentin对成纤维细胞和神经鞘细胞呈阳性表达,神经丝和髓磷脂碱性蛋白对轴突和髓磷脂鞘呈阳性表达。
1.X线摄片 可助于了解骨骼受累情况。X线平片可见各种骨骼畸形。骨内的神经纤维瘤在X线片上可显现长的条纹形态,但X线检查经常无阳性发现。 2.椎管造影、CT及MRI等检查 可发现中枢神经系统肿瘤。CT有助于了解中枢神经受累情况。良性神经纤维瘤为活跃的Ⅱ期病变,核素扫描无吸收增加,如压迫骨则有中等增加。动脉造影可见轻的新生血管反应,在晚静脉期可见肿瘤内有无血管区。大血管移位表示肿瘤起源于血管神经束部位压迫所致。 3.脑干听觉诱发电位对听神经瘤有较大诊断价值。
根据病史、临床表现、X线检查、核素扫描及动脉造影而诊断。最终依靠病理诊断。 1.定性诊断 (1)病史和家族史:本病属常染色体显性遗传、不规则遗传,仔细询问可发现家族患病者。 (2)临床特征:疣状性增生,皮下梭形的神经瘤,丛状神经瘤,色素沉着是本病四大特征。 (3)实验室检查:常染色体异常,组织病理可以确诊。 2.分型诊断 (1)1型NF:具备以下标准中的2项或多项可诊断。 ①6个或6个以上的咖啡斑,其大小为:青春期前最大直径为5cm,成人则最大直径为15cm。 ②2个或多个任何类型的神经纤维瘤或1个丛状神经纤维瘤。 ③腋部和鼠蹊部雀斑。 ④视神经胶质瘤。 ⑤2个或多个Lisch结节。 ⑥明显的骨损害如伴发或不伴发假性骨关节病的蝶部发育异常和长骨皮质变薄。 ⑦一级亲属(父母、兄弟姊妹、子女)患本病。 (2)2型NF:诊断需具备以下标准中的任一条。 ①CT或MRI:证实有双侧听神经肿瘤。 ②一级亲属中患2型神经纤维瘤或以下肿瘤,如单侧听神经瘤或以下肿瘤(神经纤维瘤、脑膜瘤、神经胶质瘤、神经鞘瘤或幼年性后囊晶状体混浊)中的2个。 3.中医 认为本病多因先天素质缺陷,或劳伤肺气,腠理不密,外邪所搏,气血不和,阻滞经络而发于肌肤。[收起]
根据病史、临床表现、X线检查、核素扫描及动脉造影而诊断。最终依靠病理诊断。 1.定性诊断 (1)病史和家族史:本病属常染色体显性遗传、不规则遗传,仔细询问可发现家族患病者。 (2)临床特征:疣状性增生,皮下梭形的神经瘤,丛状神经瘤,色素沉着是本病四大特征。 (3)实验室检查:常染色体异常,组织病理可以确诊。 2.分型诊断 (1)1型NF:具备以下标准中的2项或多项可诊断。 ①6个或6个以上的咖啡斑,其大小为:青春期前最大直径为5cm,成人则最大直径为15cm。 ②2个或多个任何类型的神经纤维瘤或1个丛状神经纤维瘤。...[详细]
手术切除为惟一的治疗方法。伴发颅内脑膜瘤和神经胶质瘤、周围神经肉瘤和其他恶性肿瘤者预后不良。 中医治则:法宜中和气血,通经活络,软坚内消。 方药:丹参15g、鸡血藤30g、赤芍15g、红花15g、厚朴10g、化橘红10g、白芥子10g、橘络10g、全丝瓜10g、白僵蚕10g、土贝母10g。
预后良好,较少发生恶变者,伴发颅内脑膜瘤和神经胶质瘤、周围神经肉瘤和其他恶性肿瘤者预后不良。本病一般发展缓慢,但青春期或妊娠期可加速发展,恶变发生率约为7%。伴发颅内脑膜瘤和神经胶质瘤、周围神经肉瘤和其他恶性肿瘤者预后不良。
进行遗传咨询。预防措施包括避免近亲结婚、携带者基因检测及产前诊断和选择性人工流产等,防止患儿出生。 散发者早期诊治,可延长存活期。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号