-
科室:
肾内科
-
别名:
巴特尔综合征
慢性特发性低钾血症
先天性醛固酮增多症
-
症状:
乏力
-
发病部位:
暂无
-
多发人群:
所有群体
-
相关疾病:
利德尔综合征
本综合征是一常染色体隐性遗传病,由Bartter(1962)首次报道,故称为巴特综合征。其临床特征为严重的低钾血症和代谢性碱中毒,伴有高肾素、高醛固酮血症、肾小球旁器增生和肥大及肾小管保钠和浓缩功能障碍,但无高血压及水肿且对外源性血管紧张素Ⅱ无反应。现认为本综合征是由离子通道基因突变引起的临床综合征。本病又称为先天性醛固酮增多症、慢性特发性低钾血症、肾小球旁器增生综合征。近年来,分子诊断学研究揭示Bartter综合征有3种不同的临床和遗传类型,即先天性Bartter综合征,典型Bartter综合征和Gitelman综合征。通常所说的Bartter综合征是指典型Bartter综合征。先天性Bartter综合征病人发现有两种基因型,Ⅰ型是由于N-K-2CL-发生失功能性基因突变所致,Ⅱ型是由于ROMK基因突变所致。典型Bartter综合征是由于CLC-kb通道基因突变所致。[收起]
本综合征是一常染色体隐性遗传病,由Bartter(1962)首次报道,故称为巴特综合征。其临床特征为严重的低钾血症和代谢性碱中毒,伴有高肾素、高醛固酮血症、肾小球旁器增生和肥大及肾小管保钠和浓缩功能障碍,但无高血压及水肿且对外源性血管紧张素Ⅱ无反应。现认为本综合征是由离子通道基因突变引起的临床综合征。本病又称为先天性醛固酮增多症、慢性特发性低钾血症、肾小球旁器增生综合征。近年来,分子诊断学研究揭示Bartter综合征有3种不同的临床和遗传类型,即先天性Bartter综合征,典型Bartter综合征和Gitelman综合征。通常所说的Bartter综合征是指典型Bartter综合征。先天性Bar...[详细]
本病病因尚无定论。多数学者认为是常染色体隐性遗传性疾病。曾有一家9个同胞中5个患病和一家连续2代4例患病的报告。现代分子生物学技术也揭示Bartter综合征是由肾小管上皮细胞上的离子转运蛋白基因突变所引起。目前已发现婴儿型Batter综合征存在Na-K-2Cl-基因突变,该基因位于15q12-21,有16个外显子,编码1099个氨基酸,为Na-K-2Cl-通道,已发现20多种突变。经典型Bartter综合征系由CICNKB基因突变所致,该基因位于1q38,编码含687个氨基酸的细胞基底侧的Cl-通道,现已发现约20种突变类型。成人型Bartter综合征又称Batter-Gietlman综合征,系由噻嗪敏感的Na-K通道基因(SCI12A3)突变所致,该基因定位于16q913,编码1021个氨基酸,已发现多达40种突变。此外还有一些病人中发现钾通道基因(ROWK)突变。因此Batter综合征可以认定为由上述几种离子通道基因突变引起的临床综合征。[收起]
本病病因尚无定论。多数学者认为是常染色体隐性遗传性疾病。曾有一家9个同胞中5个患病和一家连续2代4例患病的报告。现代分子生物学技术也揭示Bartter综合征是由肾小管上皮细胞上的离子转运蛋白基因突变所引起。目前已发现婴儿型Batter综合征存在Na-K-2Cl-基因突变,该基因位于15q12-21,有16个外显子,编码1099个氨基酸,为Na-K-2Cl-通道,已发现20多种突变。经典型Bartter综合征系由CICNKB基因突变所致,该基因位于1q38,编码含687个氨基酸的细胞基底侧的Cl-通道,现已发现约20种突变类型。成人型Bartter综合征又称Batter-Gietlman综合征,...[详细]
本症的发病机制尚未完全阐明。有人就本综合征发病环节提出4种假说: 1.血管壁对ATI的反应有缺陷导致肾素生成增多和继发性醛固酮增多。 2.近端小管钠重吸收障碍导致钠负平衡;低钠饮食亦不能逆转肾性失钾。 3.前列腺素生成过多,使肾小管失钠,血钠减低从而激活肾素-血管紧张素系统。 4.髓襻升支厚壁段对氯化物转输障碍,使氯化物重吸收减少,钾排泄增多导致低钾血症;低钾血症刺激前列腺素E2的生成,并使血浆肾素活性和血管紧张素Ⅰ升高。前列腺素E2升高后血管对ATI不敏感,因而血压正常。 近年来的临床与实验研究对Bartter综合征发病机制的认识有了很大的进展,认为Bartter综合征是由于髓襻升支厚壁段穿上皮细胞Cl-、Na的转运障碍所致。目前对髓襻升支的几种离子通道蛋白的基因编码已经克隆出来,由于这些离子通道蛋白发生了丧失功能的基因突变,致使离子转运功能发生障碍。正常肾单位髓襻升支厚壁段(图1)对Cl-、Na再吸收是由对布美他尼敏感的钠-钾-2氯运载体(bumetanide-sensitive sodium-potassium-2-chloride transporter,NKCC2)进行的。由于细胞内Na与C1-较细胞外低,NKCC2将Na、K、2Cl-运转入细胞内,仍维持电中性。上皮细胞的基侧膜上有Na-K-ATP酶能把过多的Na泵出细胞外,进入血液。另外,还有肾脏特异性基侧氯通道(kidney specific base lateral channel,CIC-kb)把Cl-泵出细胞外,经血液再吸收。髓襻升支厚壁段上的管腔膜上还有ATP调节钾通道(ATP-regulated potassium channel,ROMK)。NKCC2的转运速率是由ROMK对钾再循环进行调节,即ROMK为NKCC2提供有效的K浓度,保证管腔的正电位。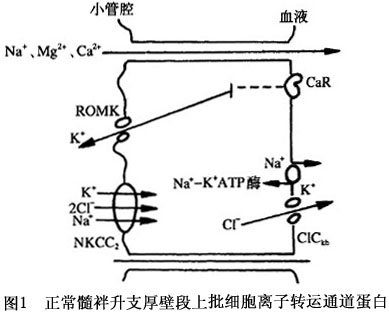 基因研究推断,上述离子运载体蛋白或通道蛋白中任何一种发生突变,都可能出现离子转运障碍,从而导致Bartter综合征的发生。不同的通道蛋白或载体的缺陷可形成Bartter综合征的不同的亚型。目前认为,由于NKCC2功能丧失性突变,导致Na、K的再吸收障碍;ClCkb通道蛋白失活,限制了NKCC2运载体的转运速率,损害了K的再循环过程对K的再吸收。所以,只要上述环节中任何一种环节上发生了功能丧失性突变,都会削减上皮细胞电位差,减少上述离子重吸收的驱动力(图2)。
基因研究推断,上述离子运载体蛋白或通道蛋白中任何一种发生突变,都可能出现离子转运障碍,从而导致Bartter综合征的发生。不同的通道蛋白或载体的缺陷可形成Bartter综合征的不同的亚型。目前认为,由于NKCC2功能丧失性突变,导致Na、K的再吸收障碍;ClCkb通道蛋白失活,限制了NKCC2运载体的转运速率,损害了K的再循环过程对K的再吸收。所以,只要上述环节中任何一种环节上发生了功能丧失性突变,都会削减上皮细胞电位差,减少上述离子重吸收的驱动力(图2)。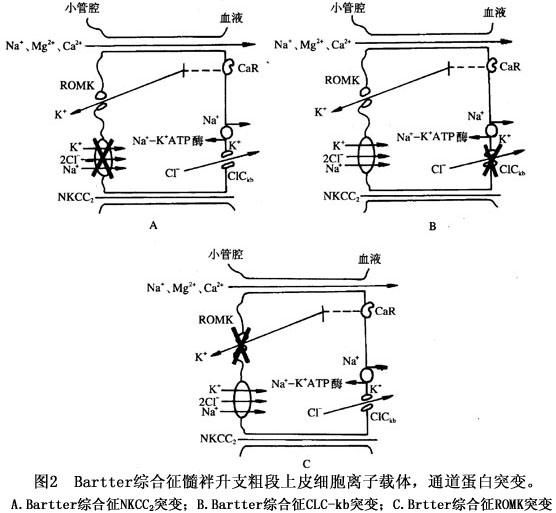 髓襻升支厚段再吸收Na、Cl-减少,细胞外液量轻度降低,继发高肾素、高醛固酮血症和肾小球旁器增生与肥大。由于氯化钠大量流经集合管,刺激泌H、泌K,加上高醛固酮血症,因而引起低钾血症和代谢性碱中毒。肾素-血管紧张素-醛固酮系统功能亢进,促进激肽、血管舒缓素生成,前列腺素生成增多,使血管对血管紧张素反应降低,血压保持正常,无水肿表现。最近研究发现,Bartter综合征患者单核细胞NO合成酶(ecNOS)mRNA水平呈高表达,尿中NO代谢产物NO2-/NO3-与cGMP平行升高,推测由于NO产生增多,减少血管张力,认为也是Bartter综合征患者血管对血管紧张素反应性降低的原因之一。有关ecNOS在Bartter综合征发病机制中的作用,尚需深入研究。[收起]
本症的发病机制尚未完全阐明。有人就本综合征发病环节提出4种假说: 1.血管壁对ATI的反应有缺陷导致肾素生成增多和继发性醛固酮增多。 2.近端小管钠重吸收障碍导致钠负平衡;低钠饮食亦不能逆转肾性失钾。 3.前列腺素生成过多,使肾小管失钠,血钠减低从而激活肾素-血管紧张素系统。 4.髓襻升支厚壁段对氯化物转输障碍,使氯化物重吸收减少,钾排泄增多导致低钾血症;低钾血症刺激前列腺素E2的生成,并使血浆肾素活性和血管紧张素Ⅰ升高。前列腺素E2升高后血管对ATI不敏感,因而血压正常。 近年来的临床与实验研究对Bartter综合征发病机制的认识有了很大的进...[详细]
髓襻升支厚段再吸收Na、Cl-减少,细胞外液量轻度降低,继发高肾素、高醛固酮血症和肾小球旁器增生与肥大。由于氯化钠大量流经集合管,刺激泌H、泌K,加上高醛固酮血症,因而引起低钾血症和代谢性碱中毒。肾素-血管紧张素-醛固酮系统功能亢进,促进激肽、血管舒缓素生成,前列腺素生成增多,使血管对血管紧张素反应降低,血压保持正常,无水肿表现。最近研究发现,Bartter综合征患者单核细胞NO合成酶(ecNOS)mRNA水平呈高表达,尿中NO代谢产物NO2-/NO3-与cGMP平行升高,推测由于NO产生增多,减少血管张力,认为也是Bartter综合征患者血管对血管紧张素反应性降低的原因之一。有关ecNOS在Bartter综合征发病机制中的作用,尚需深入研究。[收起]
本症的发病机制尚未完全阐明。有人就本综合征发病环节提出4种假说: 1.血管壁对ATI的反应有缺陷导致肾素生成增多和继发性醛固酮增多。 2.近端小管钠重吸收障碍导致钠负平衡;低钠饮食亦不能逆转肾性失钾。 3.前列腺素生成过多,使肾小管失钠,血钠减低从而激活肾素-血管紧张素系统。 4.髓襻升支厚壁段对氯化物转输障碍,使氯化物重吸收减少,钾排泄增多导致低钾血症;低钾血症刺激前列腺素E2的生成,并使血浆肾素活性和血管紧张素Ⅰ升高。前列腺素E2升高后血管对ATI不敏感,因而血压正常。 近年来的临床与实验研究对Bartter综合征发病机制的认识有了很大的进...[详细]
本病临床表现呈多样化,临床类型不一,发病以青少年多见,性别无显著性差异,无种族差异。如果提高对本病的认识,临床上并不一定少见,由于合并症及并发症的出现,往往临床不易及时准确的诊断。 1.水盐代谢失常型 最多见,突出表现为低血钾性碱中毒。患者来诊的主要原因是低血钾及碱中毒,其临床表现为:疲乏无力,下肢或周身软瘫呈周期性瘫痪现象;感觉迟钝,心律失常,腹胀,肠麻痹,肠梗阻,恶心,呕吐,排尿困难,晕厥,神智障碍,反射迟钝,腱反射减弱或消失等低血钾症状;持续低血钾可发生糖代谢紊乱,糖耐量减低,胰岛素释放受影响,脑电图有异常波型。血钾<3.0mmol/L,尿钾>50mmol/24h以上。碱中毒与低血钾经常同时发生,有手足麻木,抽搐,呼吸气短,精神兴奋或躁动,肌肉颤抖及腹痛等症,Chvostek及Trosseau征阳性,血pH值>7.45,血浆:HCO3-常>24mEg/L,尿呈碱性反应。早期病人尿量增多,可达每天5000毫升以上,比重降低,尿渗透压降低,患者虽有抽搐,但血钙、磷、AKP,尿钙均可正常。 由于脱水失盐,患者经常口干、口渴、嗜盐、多饮、多尿、夜尿多、消瘦、体重减轻、便秘、皮肤弹性差、眼窝深陷、眼压低,脱水较严重时尿少,每天仅300~400ml,可发生虚脱、神志障碍或昏迷。血钠<130mmol/L,血氯<90mmol/L,尿钠、尿氯排出增加,有效血容量减少,远曲小管和球旁器进一步发生变化,引起肾素、前列腺素、血管紧张素及醛固酮分泌增多。 Zipser报道两例本病,其中1例有严重低血镁,作者认为低血镁也可兴奋肾脏PG增多而引起巴特综合征,或是另有原因,需进一步研究。 2.以肾脏病为主要临床表现类型 不少见,本病可常有肾盂肾炎,间质性肾炎,失盐性肾炎,肾小球肾炎合并肾钙化,肾结石,肾盂积水,肾功能减退等表现。由于慢性肾脏病变迁延不愈,可发生肾性骨病,骨质疏松,牙脱落,继发性甲状旁腺功能亢进等表现。并可有尿磷增多及糖尿现象。Meget报道一组巴特综合征病患者,由于肾功能异常变化而发生尿酸盐代谢异常,尿酸清除率下降,尿中尿酸盐排出减少,血液尿酸水平升高,50%患者发生高尿酸血症,20%患者发生急性痛风性关节炎。正常人痛风病发生率仅为0.2%~0.3%,而巴特综合征病人合并痛风症大大增加,痛风症也可成为巴特综合征的临床表现之一。 MeCrldie报道4例本病,其中3例有高尿钙症。巴特综合征合并肾钙化,肾结石,高尿钙症并不少见。结石性质可为草酸钙、磷酸钙、尿酸盐或为混合性。血清尿酸值>7.0mg/dl者为高尿酸血症。尿中尿酸正常值为0.5~0.8g/24h,正常尿酸清除率为6~12ml/min,而巴特综合征时排出减少。尿钙值各地区差异较大,一般来说如高于200~250mg/24h,即为高尿钙,应寻找尿钙增高原因。 3.血管活性激素平衡失调表现 巴特综合征有高前列腺素,肾素,血管紧张素和醛固酮,其血尿PGA2,PGE,PGF,PGI。都可升高,但主要是PGE升高。PGA2、PGE及PGF增高均可用阿司匹林治疗,3个月后恢复正常水平。Bowden报道7例中5例的PGE增高,用吲哚美辛治疗后4例PGE下降,排钠与排钾减少,血钾回升,血浆肾素值下降,肌酐清除率降低。巴特综合征的尿PGE和血管舒缓素排出量有关,高肾素血症是继发于肾脏PG的增加。血管舒缓素-激肽系统和前列腺素-肾素-血管紧张素-醛固酮系统有关。血管舒缓素-激肽系统活性增高,可刺激肾脏合成PGE增多,用吲哚美辛治疗后,PGE、血管舒缓素、血浆肾素活性均可明显降低,并可使AngⅡ增加敏感性,血钾恢复正常。本症时AngI也有增高,可达90~200ng/ml,而正常值仅为50ng/ml以下水平。用吲哚美辛后不能使尿Aldo排出量降低,理由不清。动物实验证实由肾动脉注入PGE和花生四烯酸后,可增加血浆肾素活性,用吲哚美辛后可增加AngⅡ的敏感性,也可降低肾素活性。Fujita给本病患者作血管紧张素注入实验,确实发现对血管紧张素的反应比正常人低下,但应用白蛋白静脉注射后就会提高反应性,说明血管壁对血管紧张素的抗压反应是因低钠、低血容量等而引起。Inada给本病患者每天入钠175mmol,并以高于20ng/(kg·min)的AngⅡ注入时,舒张压可升高20mmHg,而收缩压要大于100ng/(kg·min)的注入速度时,才有轻微上升,而正常人仅仅在注入20ng/(kg·min)的速度即能提高收缩压20mmHg,舒张压20mmHg,显然巴特征患者对外源性的AngⅡ反应性不敏感。 巴特综合征的PG增高是原发性的,而血浆肾素活性,血管紧张素及醛固酮增高均为继发性的反应。正常血浆Aldo值为5.0~15.0ng/dl而巴特综合征患者可达50ng/dl以上,尿Aldo值正常为5.0~20.0ng/24h,而巴特综合征可达30ng/24h以上或更高。 4.其他临床表现 儿童时期发病者常有生长发育障碍,生长停滞或缓慢,智力落后及性腺功能低下,但未见垂体侏儒症表现。巴特综合征病人肾功能减退时可合并贫血。脱水较严重时可伴有血液浓缩,血红蛋白达16克以上,并伴有红细胞增多症等。[收起]
本病临床表现呈多样化,临床类型不一,发病以青少年多见,性别无显著性差异,无种族差异。如果提高对本病的认识,临床上并不一定少见,由于合并症及并发症的出现,往往临床不易及时准确的诊断。 1.水盐代谢失常型 最多见,突出表现为低血钾性碱中毒。患者来诊的主要原因是低血钾及碱中毒,其临床表现为:疲乏无力,下肢或周身软瘫呈周期性瘫痪现象;感觉迟钝,心律失常,腹胀,肠麻痹,肠梗阻,恶心,呕吐,排尿困难,晕厥,神智障碍,反射迟钝,腱反射减弱或消失等低血钾症状;持续低血钾可发生糖代谢紊乱,糖耐量减低,胰岛素释放受影响,脑电图有异常波型。血钾<3.0mmol/L,尿钾>50mmol/24h以上。碱中毒...[详细]
巴特综合征病人可合并痛风症。合并肾钙化,肾结石,高尿钙症。巴特综合征病人肾功能减退时可合并贫血。
1.血钾、钠、氯多低于正常水平。 2.血pH值可高于7.46呈碱血症,C02CP高于30mmol/L以上。 3.血浆肾素活性(PRA)增高可达(4.5±2.9)μg/L·h以上。 4.血醛固酮(Aldo)值升高可达101±9ng/L。 5.血中PGA、PGE、PGF、PGI均可升高,如PGF可达(138.0±78.0)ng/ml。 6.血管紧张素Ⅱ(AngⅡ)升高可达(95.8±35.2)ng/L。 7.肾功能检查 BUN可升高达7.0mmol/L以上,毛森试验比重低,夜尿量增多,并可发现尿中有蛋白及红白细胞等。 8.肾功能减退后期可发现血钙降低,血磷上升,AKP升高,尿酸升高,肌酐升高,PTH升高,有继发甲旁亢表现。 9.尿17-OHCS,尿17-KS多在正常范围。[收起]
1.血钾、钠、氯多低于正常水平。 2.血pH值可高于7.46呈碱血症,C02CP高于30mmol/L以上。 3.血浆肾素活性(PRA)增高可达(4.5±2.9)μg/L·h以上。 4.血醛固酮(Aldo)值升高可达101±9ng/L。 5.血中PGA、PGE、PGF、PGI均可升高,如PGF可达(138.0±78.0)ng/ml。 6.血管紧张素Ⅱ(AngⅡ)升高可达(95.8±35.2)ng/L。 7.肾功能检查 BUN可升高达7.0mmol/L以上,毛森试验比重低,夜尿量增多,并可发现尿中有蛋白及红白细胞等。 8.肾功...[详细]
1.静脉肾盂造影 可发现肾结石,肾盂积水等异常。 2.心电图 可发现低血钾表现。 3.肾图异常。 4.肾活检 肾小球旁器增生肥大可在肾活检时发现,有球旁细胞、致密斑细胞、极周细胞及球外系膜细胞的数量增多,或细胞呈肥大分泌现象。95%肾素由球旁细胞分泌,1%~5%可由致密斑细胞、间质细胞或出球小动脉内皮细胞产生肾素。
巴特综合征的诊断主要根据有以下几点: 1.临床上有低钾血症表现,如软弱无力、周期性瘫痪、夜尿增多、心电图上有低钾表现。儿童患者尚有身高不长和智力低下。 2.碱中毒,表现为手足搐搦。 3.血钾、钠和氯化物降低。 4.血浆肾素活性,血和24h尿醛固酮增高。 5.对血管紧张素Ⅱ和血管加压素无血压升高反应。 6.肾活检有肾小球球旁器的颗粒细胞增生。 7.血压正常。 8.对先天性者,可用分子生物学技术检查基因突变。
巴特综合征的治疗初期,主要是针对低血钾及碱血症给以对症治疗,但疗效欠佳。继而针对高肾素血症,高醛固酮症给以治疗,仅部分有效。近些年来开展对高前列腺素分泌治疗,并辅以低血钾对症治疗取得了进展,但仍有报道不完全令人满意的。 补钾是必需的措施,但单独补钾,血钾不能恢复至正常水平时,应加用抗醛固酮药物,如螺内酯(安体舒通)或氨苯蝶啶,可改善疗效,一般用量为60~l80mg/d,大量可引起男性乳房增大,因此,剂量不宜过大。Solomon及Modlinger等用普萘洛尔(心得安)治疗本征,针对高肾素血症,其疗效也不满意,但普萘洛尔(心得安)合用螺内酯(安体舒通)时可改善疗效,且可恢复血钾水平。Norby等用阿司匹林,每天每公斤体重给予100mg,治疗10周后可取得满意疗效,但停药4天后又回到治疗前水平。说明阿司匹林可抑制PG合成,为有效药物,副作用小比较安全,可惜作用不持久。以后采用前列腺素合成酶抑制剂吲哚美辛(消炎痛)等药物治疗,均可减少PG合成,降低肾素、血管紧张素及醛固酮的活性,使症状改善。吲哚美辛(消炎痛)可单用,也可合用螺内酯(安体舒通),但长期应用可有钠水潴留,引起水肿或心力衰竭,因此不宜长期大量应用,主张间断应用。McGredie用二磷酸盐治疗巴特征的肾钙化,取得一定疗效。如有痛风症可用别嘌醇(吡唑嘧啶醇),或秋水仙碱等治疗,可缓解痛风性关节炎。脱水失盐患者可补给氯化钠液,低镁者可补给镁盐治疗等。肾功能减退较重者应给予透析治疗。缺钙者应补充钙剂及活性维生素D剂治疗等。[收起]
巴特综合征的治疗初期,主要是针对低血钾及碱血症给以对症治疗,但疗效欠佳。继而针对高肾素血症,高醛固酮症给以治疗,仅部分有效。近些年来开展对高前列腺素分泌治疗,并辅以低血钾对症治疗取得了进展,但仍有报道不完全令人满意的。 补钾是必需的措施,但单独补钾,血钾不能恢复至正常水平时,应加用抗醛固酮药物,如螺内酯(安体舒通)或氨苯蝶啶,可改善疗效,一般用量为60~l80mg/d,大量可引起男性乳房增大,因此,剂量不宜过大。Solomon及Modlinger等用普萘洛尔(心得安)治疗本征,针对高肾素血症,其疗效也不满意,但普萘洛尔(心得安)合用螺内酯(安体舒通)时可改善疗效,且可恢复血钾水平。N...[详细]
本病经过治疗后可得到短期缓解,但其远期疗效不佳,主要是因为患者的慢性肾功能衰竭,可发展为尿毒症而亡,或因体质差,可合并多种疾病而走向慢性过程,失去劳动力而致残。患者得病后可生存10~20年以上。
本病无有效预防措施,主要应预防慢性肾炎、间质性肾炎、肾盂肾炎等疾病,增强体质,提高免疫力,并提高对本病的认识,早期诊断,早期治疗。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号