-
科室:
内分泌科
-
别名:
syndrome of thyroid hormone resistance
syndrome of resistance to thyroid hormone
甲状腺激素不应症
-
症状:
心悸
生长缓慢
-
发病部位:
暂无
-
多发人群:
儿童、青少年人群
-
相关疾病:
暂无
甲状腺激素抵抗综合征(Thyroid hormone resistance syndrome,SRTH)也称甲状腺激素不应症或甲状腺激素不敏感综合征(thyroid hormone insensitivity syndrome,THIS),由Refetoff在1967年首次报道。本病以家族性发病为多见,也有少数为散发病例,约占1/3。大都在儿童和青少年发病,年龄最小的为新生儿,男女性别均可患病。临床表现血清游离T4 (FT4)和游离T3 (FT3)持续升高,同时促甲状腺激素(TSH)正常,病人没有药物、非甲状腺疾病和甲状腺激素转运异常的影响。最特异的表现是给以病人超生理剂量甲状腺激素后,不能抑制升高的TSH下降到正常水平,同时也没有外周组织对过量甲状腺激素的反应。 其病因包括甲状腺激素受体突变或甲状腺激素和受体结合障碍或甲状腺激素受体结合后作用异常等原因,导致组织器官对甲状腺激素反应减低,引起代谢和甲状腺功能异常等表现。全身除了脑、睾丸、淋巴器官外,其他器官、组织和细胞都有甲状腺激素受体。临床上多见的是部分抵抗,真正完全性抵抗是很少见的,而各个器官、组织对甲状腺激素抵抗程度不同,病人的代偿能力不同,所以临床有不同的表现和实验室特征。 甲状腺激素抵抗有几种情况,最常见的为垂体抵抗和全身抵抗,临床可表现甲状腺功能亢进(甲亢)、甲状腺功能正常或甲状腺功能低减(甲减)。如果垂体和周围组织对甲状腺激素的抵抗是相似的,病人表现为甲状腺功能正常;如果垂体抵抗低于周围抵抗,病人表现甲减;如果垂体抵抗高于周围抵抗,病人表现甲亢。甲状腺激素抵抗病人常常被偶然发现,可被不恰当地处理,如甲状腺切除、核素治疗或硫脲类药物治疗,虽然部分患者血清TSH正常,但由于同时血清甲状腺激素升高,所以TSH仍然为不恰当升高。[收起]
甲状腺激素抵抗综合征(Thyroid hormone resistance syndrome,SRTH)也称甲状腺激素不应症或甲状腺激素不敏感综合征(thyroid hormone insensitivity syndrome,THIS),由Refetoff在1967年首次报道。本病以家族性发病为多见,也有少数为散发病例,约占1/3。大都在儿童和青少年发病,年龄最小的为新生儿,男女性别均可患病。临床表现血清游离T4 (FT4)和游离T3 (FT3)持续升高,同时促甲状腺激素(TSH)正常,病人没有药物、非甲状腺疾病和甲状腺激素转运异常的影响。最特异的表现是给以病人超生理剂量甲状腺激素后,不能抑...[详细]
SRTH的确切病因不清楚,绝大多数是由于甲状腺激素受体基因发生突变。最常见的是甲状腺激素受体基因核苷酸发生变化或者缺失,使甲状腺激素受体的氨基酸顺序发生变化,导致受体结构和功能的变化,对甲状腺激素发生抵抗或不敏感。其次为甲状腺激素受体数目减少,导致甲状腺激素作用减弱。还有甲状腺激素受体后作用发生障碍,也可引起SRTH。
最常见的为甲状腺激素受体(c-erbAβ)β型功能区配体结合缺陷,基因位于第3号染色体的短臂上;其次为甲状腺激素受体亲和力减低。由于抵抗程度不同,临床表现不同,哪个器官对甲状腺激素敏感,那个器官的临床表现就敏感突出。如果心脏对甲状腺激素抵抗较轻,病人表现心动过速。 甲状腺激素抵抗主要是T3核受体缺陷,体外培养的淋巴母细胞也表现对甲状腺激素抵抗,研究证明患者外周血淋巴细胞T3核受体和T3的亲和力只为正常对照组的1/10;也有作者证明患者淋巴细胞结合甲状腺激素的Ka值是正常的,但结合容量减低;还有的患者淋巴细胞T3核受体正常,但其他组织如垂体、肝脏、肾脏、心脏存在T3核受体缺陷。 甲状腺激素受体TR-α和TR-β基因分别位于第17染色体和第3染色体上。全身性SRTH的研究发现,T3核受体区的β基因发生了点突变,即甲状腺激素受体β基因中有一个核苷酸被另一个核苷酸代替,从而导致甲状腺激素受体中相应位置的氨基酸被另一个氨基酸取代,使受体功能表现异常;或者数个碱基对缺失;或者单个核苷酸缺失;或者核苷酸插入;或者发生数个碱基复制等等。点突变出现在T3核受体和T3结合区的中部及羟基端,导致激素和受体亲和力减低。患者多为杂合子,即只要有一条T3核受体β等位基因点突变即可发病,属于常染色体显性遗传。也有少数全身激素抵抗的患者T3核受体β基因有大片丢失,即甲状腺激素受体基因中一个编码,氨基酸密码子突变为终止密码子,使表达的甲状腺激素受体过早终止密码子,导致甲状腺激素受体丢失了部分氨基酸,这种氨基酸缺失可以是单个,也可以是多个。出现在受体DNA结合区及T3结合区上,病人均表现纯合子,即必须两条等位基因同时发生基因缺失才会发病,遗传方式为常染色体隐性遗传。临床上表现为多样性,可能因为基因突变或缺失的多变性,而不是受体数量减少的多样性。而甲状腺激素受体α基因突变很少有报道。 对选择性垂体抵抗患者也发现有T3核受体β2基因突变,这种基因只分布在垂体和一些神经组织中,所以临床仅仅表现垂体抵抗;另一种原因是垂体组织中使T4脱碘生成R的特异性Ⅱ型-5’脱碘酶有缺陷,表现垂体组织抵抗。 镜下染色体没有发现异常,异常发生在分子DNA水平,总之SRTH发病机制是在分子水平上,是一种典型的受体病。 很少有关于SRTH病人的病理性改变,从一例病人肌肉活检,电镜下发现线粒体肿胀,和甲亢相似。用甲苯胺蓝染色皮肤成纤维细胞,光镜下发现中度至重度异染粒,在甲减黏液性水肿皮肤也有这种细胞外异染物质沉积,在SRTH中这种表现可能是皮肤组织甲状腺激素作用降低引起,甲状腺激素治疗并不能使SRTH病人成纤维细胞的异染粒消失。从活检或外科手术取得病人的甲状腺组织,见到滤泡上皮有不同程度的增生,大小不等,有些病人呈现腺瘤样甲状腺肿,或者胶质样甲状腺肿,或者正常的甲状腺组织。[收起]
最常见的为甲状腺激素受体(c-erbAβ)β型功能区配体结合缺陷,基因位于第3号染色体的短臂上;其次为甲状腺激素受体亲和力减低。由于抵抗程度不同,临床表现不同,哪个器官对甲状腺激素敏感,那个器官的临床表现就敏感突出。如果心脏对甲状腺激素抵抗较轻,病人表现心动过速。 甲状腺激素抵抗主要是T3核受体缺陷,体外培养的淋巴母细胞也表现对甲状腺激素抵抗,研究证明患者外周血淋巴细胞T3核受体和T3的亲和力只为正常对照组的1/10;也有作者证明患者淋巴细胞结合甲状腺激素的Ka值是正常的,但结合容量减低;还有的患者淋巴细胞T3核受体正常,但其他组织如垂体、肝脏、肾脏、心脏存在T3核受体缺陷。...[详细]
本病多发生于青少年及儿童,男女发病比率为1.2∶1,根据其发病及临床表现可分为3种类型。 1.全身性甲状腺激素不应症 垂体与周围组织均受累,本型又可分为甲状腺功能代偿性正常型及甲状腺功能减退症型。 (1)代偿性正常型:多为家族性发病,少数为散发者,本型发病多较轻。家系调查多为非近亲婚配,属常染色体显性遗传。本型患者的垂体及周围组织对甲状腺激素抵抗或不敏感程度较轻,甲状腺功能状态表现被高T3、T4代偿,可维持正常的状态,无甲亢临床表现,智力正常,无耳聋,无骨骺愈合发育延迟,但可有不同程度的甲状腺肿及骨化中心延迟表现,其血中甲状腺激素浓度(T3、T4、FT3、FT4)均有升高,TSH值升高或正常,TSH不受高T3及T4的抑制。 (2)甲状腺功能减退型:本型特点为血中甲状腺激素水平升高,而临床表现甲减,多属常染色体隐性遗传。本型可表现为智力差,发育落后,可有骨成熟落后表现,有点彩样骨骼,骨龄落后,还可有翼状肩胛、脊柱畸形、鸡胸、鸟样颜面、舟状颅及第四掌骨变短等异常表现。有些患者尚可发生先天性聋哑、少动、缄默及眼球震颤等异常。查体可有甲状腺肿,血中T3、T4、FT3及FT4水平升高,TSH分泌不受T3抑制,TSH对TRH反应增强。本型甲减与呆小病及黏液性水肿表现有区别。 2.选择性垂体对甲状腺激素不应症 本型特点为垂体多有受累,对甲状腺激素不反应,而其余外周组织均不受累,可对甲状腺激素反应正常,其临床表现有甲亢,但TSH水平亦高于正常,而又无垂体分泌TSH瘤的存在。本型又可分为以下2型。 (1)自主型:根据TSH对TRH及T3、T4的反应性,本型TSH升高,垂体TSH对TRH无明显反应,高水平的T3、T4仅轻微抑制TSH分泌,地塞米松也只轻微降低TSH分泌,故称自主型,但无垂体瘤存在。患者有甲状腺肿及甲亢临床表现,但无神经性耳聋,骨骺可愈合延迟,还可无身材矮小,智力差,计算力差及其他骨发育异常。 (2)部分型:临床表现可同自主型,但又不及自主型明显,临床表现可有甲亢,且TSH升高,垂体TSH对TRH、T3有反应性,但其反应性又可部分被T3及T4所抑制。本型还可有胱氨酸尿症。 3.选择性外周组织对甲状腺激素不应症 本型特点为周围组织对甲状腺激素不反应或不敏感,而垂体多无受累,对甲状腺激素正常反应。临床表现甲状腺肿大,无聋哑及骨骺变化,虽甲状腺激素正常及TSH正常,但临床有甲状腺功能低下表现,心动过缓,水肿、乏力、腹胀及便秘等异常,本型患者给予较大剂量的甲状腺制剂后可获病情缓解,因为其甲状腺功能及TSH正常水平,因此临床上对本型患者常常漏诊或误诊。[收起]
本病多发生于青少年及儿童,男女发病比率为1.2∶1,根据其发病及临床表现可分为3种类型。 1.全身性甲状腺激素不应症 垂体与周围组织均受累,本型又可分为甲状腺功能代偿性正常型及甲状腺功能减退症型。 (1)代偿性正常型:多为家族性发病,少数为散发者,本型发病多较轻。家系调查多为非近亲婚配,属常染色体显性遗传。本型患者的垂体及周围组织对甲状腺激素抵抗或不敏感程度较轻,甲状腺功能状态表现被高T3、T4代偿,可维持正常的状态,无甲亢临床表现,智力正常,无耳聋,无骨骺愈合发育延迟,但可有不同程度的甲状腺肿及骨化中心延迟表现,其血中甲状腺激素浓度(T3、T4、FT3、FT4)均有升高...[详细]
1986年用分子生物学方法克隆出核T3受体(TRs),此后有关TRs的研究迅速进展,并对发病机制作出进一步解释。本病与TRs缺陷有关,其缺陷表现形式有多样,并推测本病可能存在着两种TRs,其中异常的受体可抑制核T3受体复合物与染色质DNA的合成。患者淋巴细胞结合甲状腺激素的Ta值正常,但结合容量下降,提示家族性生化缺陷可能是TRs蛋白的缺乏。有些患者不存在淋巴细胞或成纤维细胞。TRs的异常,但不排除本病患者的其他靶腺组织,如垂体、肝、肾、心、皮肤等有TRs的缺陷。还有可能是缺陷不在受体水平,而是在受体后水平。目前研究已进入了基因水平,其发病机制与分子缺陷和突变本质有关,如全身性甲状腺激素不应症发病较多,此型患者的受体基因改变出现在TRβ上,尚未发现TRα基因异常,说明一条等位基因的点突变就可引起本病。目前认为本病多因TRs基因表达的多方面失调所致,它是发生在受体分子水平上,并且是一种典型的受体疾病。因此,实验室检查对本病的诊断相当重要,并要求有分子生物学实验室条件。 1.放免检测甲状腺功能 T3、T4、FT3、FT4、TSH、TBG、TRH兴奋试验等。T3、T4可结构正常,有免疫活性,其值常常超过正常3倍多。 2.PBI值升高,BMR正常,过氯酸盐试验阴性,131Ⅰ吸碘率正常或升高。 3.血中LATS阴性,TGA(-)、TMA(-)。 4.染色体测定可发现异常。 5.DNA、核T3受体(TRs)、基因TRβ、TRα检测,TRβ基因发生点突变,碱基替换多出现在TRβ的T3结合区的中部及羟基端,即外显子6、7、8上,导致受体与T3亲和力下降。少数患者属常染色体隐性遗传者,基因分析发现TRβ基因大片缺失,出现受体DNA结合区及T3结合区上,病人均为纯合子,而仅有一条TRβ等位基因缺失的杂合子家族成员不发病。[收起]
1986年用分子生物学方法克隆出核T3受体(TRs),此后有关TRs的研究迅速进展,并对发病机制作出进一步解释。本病与TRs缺陷有关,其缺陷表现形式有多样,并推测本病可能存在着两种TRs,其中异常的受体可抑制核T3受体复合物与染色质DNA的合成。患者淋巴细胞结合甲状腺激素的Ta值正常,但结合容量下降,提示家族性生化缺陷可能是TRs蛋白的缺乏。有些患者不存在淋巴细胞或成纤维细胞。TRs的异常,但不排除本病患者的其他靶腺组织,如垂体、肝、肾、心、皮肤等有TRs的缺陷。还有可能是缺陷不在受体水平,而是在受体后水平。目前研究已进入了基因水平,其发病机制与分子缺陷和突变本质有关,如全身性甲状腺激素不应症...[详细]
1.X线骨骺检查 多有骨骺发育延迟,点彩状骨骺和其他骨骺畸形。 2.甲状腺B超检查 了解甲状腺肿大程度,有无结节等。 3.其他测定 如尿胱氨酸测定,5’-脱碘酶等生化检测等。
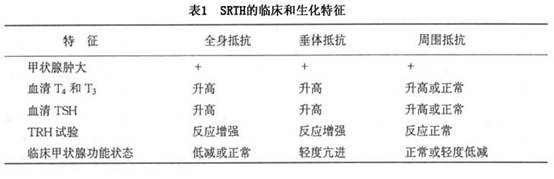
本病临床表现复杂以及一般医院检查条件所限或认识不足,因此,诊断常有延误或漏诊。诊断本病时患者的甲状腺肿多为Ⅰo或Ⅱo ,血清T3、T4水平升高,而临床表现甲状腺功能正常或有甲减时,均应考虑本病的可能性。如同时伴有家族性发病,TSH水平升高或正常、智力低下,骨骺成熟延缓,点彩状骨骼,先天性聋哑,过氯酸盐试验阴性及TGA及TMA阴性等,则为较典型的甲状腺激素不应症(表1)。
甲状腺激素不应症临床表现有所区别,故治疗不同,未来可采用基因治疗,目前常用方法如下: 1.抗甲状腺药物治疗 已知本病并不是由于甲状腺激素水平升高所致,而是受体(核T3受体)对甲状腺激素不敏感,血中甲状腺激素水平升高,并具有代偿意义。使用抗甲状腺药物人为地降低血中T3、T4水平,可能加重甲减表现,促进甲状腺肿加重,并促进TSH分泌增多与垂体分泌TSH细胞增生与肥大,尤其是儿童甲减对生长发育不利,所以不主张采用抗甲状腺药物治疗。只有对部分靶器官不反应型患者。可在观察下试用抗甲状腺药物治疗,如疗效不佳,及时停用。 2.甲状腺激素治疗 可根据病情与类型应用及调整,全身性甲状腺激素不应症患者一般不需甲状腺素治疗,甲减型可采用T4及碘塞罗宁(T3)治疗,尤其是对婴幼儿及青少年有益,可促进生长发育,缩小甲状腺肿及减少TSH分泌,一般采用左甲状腺素钠(L-T4)片,2次/d,每次100~200μg,应用T3制剂也有疗效。对于外周组织的甲状腺激素不应症应给予较大剂量的甲状腺制剂可使病情好转。对于垂体性的甲状腺激素不应症应控制甲亢症状,可应用抗甲状腺药物或131Ⅰ治疗等。 3.糖皮质激素治疗 糖皮质激素可减少TSH对TRH的兴奋反应,但甲状腺激素不应症患者是否有反应,尚无统一意见,有人采用地塞米松,4次/d,每次2~3mg,溴隐亭每天2.5mg及左甲状腺素钠(L-T4)片,5次/d,每次2mg,发现疗效甚好,但不宜长期应用,地塞米松的副作用较大。 4.多巴胺激动药 1984年,BaJorunas等报告应用溴隐亭治疗一例男性成人甲状腺激素不应症,开始剂量为每天2.5mg,渐增至每天10mg,疗程16个月,于用药7个月时其TSH水平下降,TSH及PRL对TRH的反应值下降,T4及T3水平升高,继续用药后其T4及T3水平下降,131Ⅰ吸碘率也下降,甲状腺肿缩小,但停用溴隐亭后4个月又复发。也可试用其他种类的多巴胺能激动药,但疗效也有待观察肯定。 5.生长抑素等 可选用本药抑制TSH分泌,及三碘甲腺酐酸(triiodothyroacetic acid)和多巴胺等均可抑制TSH分泌。三碘甲腺酐酸结构与碘塞罗宁(T3)相似,对垂体有负反馈作用,但无升高代谢的副作用。此外,可给予普萘洛尔(心得安)每天30~60mg,有助于减轻临床症状。 6.基因治疗 清楚发病机制后,可开展基因治疗与受体病治疗。[收起]
甲状腺激素不应症临床表现有所区别,故治疗不同,未来可采用基因治疗,目前常用方法如下: 1.抗甲状腺药物治疗 已知本病并不是由于甲状腺激素水平升高所致,而是受体(核T3受体)对甲状腺激素不敏感,血中甲状腺激素水平升高,并具有代偿意义。使用抗甲状腺药物人为地降低血中T3、T4水平,可能加重甲减表现,促进甲状腺肿加重,并促进TSH分泌增多与垂体分泌TSH细胞增生与肥大,尤其是儿童甲减对生长发育不利,所以不主张采用抗甲状腺药物治疗。只有对部分靶器官不反应型患者。可在观察下试用抗甲状腺药物治疗,如疗效不佳,及时停用。 2.甲状腺激素治疗 可根据病情与类型应用及调整,全身性甲状腺激...[详细]
甲状腺激素不应症是遗传性受体疾病,目前尚无特效治疗方法,由于其临床分类不同,治疗反应多不一致,大多数临床学家普遍认为垂体性甲状腺激素不应症的疗效较好。而部分靶组织对甲状腺激素不应症的治疗较困难,且本病早期诊断多有困难,故对新生儿有家族史者应进行全面检查,尤其是对智力低下,聋哑和体型异常的患者更应注意。
本病属常染色体显性遗传,对于育龄妇女有家族史者应进行教育,最好是计划生育或节育。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号