遗传性球形红细胞增多症(hereditary spherocytosis,HS)是一种先天性红细胞膜骨架蛋白异常引起的遗传性溶血病。其主要特点是外周血中见到较多小球形红细胞。临床上以贫血、黄疸、脾肿大、血液中球形红细胞增多、病程呈慢性贫血经过并伴有溶血反复急性发作为主要特征。现已明确,HS是一种红细胞膜蛋白基因异常引起的遗传性疾病。
本病溶血的主要原因是先天性红细胞膜蛋白基因突变导致的红细胞膜异常。 HS的分子遗传学异常主要包括锚蛋白和膜收缩蛋白联合缺乏、带3蛋白缺乏、单纯膜收缩蛋白部分缺乏和4.2蛋白缺乏,以锚蛋白和膜收缩蛋白联合缺乏最常见。上述膜蛋白异常可导致膜骨架与膜之间的垂直方向相互作用减弱,从而使膜脂质双层变得不稳定,部分脂质以出芽形式形成囊泡而丢失,红细胞膜表面积减少,最终使红细胞形成小球形。另外,HS红细胞(特别是经过脾脏的红细胞)都有一定程度的脱水和对单价离子通透性异常,这可能也与膜骨架缺陷有关。 由于球形细胞内容积储备很低,其变形性能因而降低,难于通过直径比其本身小得多的脾微循环而阻留于脾髓内被吞噬和清除。在脾脏,还可能由于红细胞被阻留于脾髓内的时间长、红细胞ATP生成不足、pH值下降,使红细胞更易变为球形。此外,由于本病红细胞内的ATP相对缺乏,使红细胞的除钙作用减弱,钙沉积于细胞膜上而使膜变硬,因而在脾内更易于破碎。未破坏的红细胞多次经过脾循环后,其脆性进一步增加,球形更明显,在脾内易于破坏。 实验证明,脾切除后贫血的纠正程度与红细胞膜收缩蛋白的原有缺乏程度有关。收缩蛋白>正常70%者,术后贫血可完全纠正;为正常40%~70%者,可得到代偿;<正常40%者术后仍有贫血。所幸<40%的病例常为隐性遗传患者,临床少见。[收起]
本病溶血的主要原因是先天性红细胞膜蛋白基因突变导致的红细胞膜异常。 HS的分子遗传学异常主要包括锚蛋白和膜收缩蛋白联合缺乏、带3蛋白缺乏、单纯膜收缩蛋白部分缺乏和4.2蛋白缺乏,以锚蛋白和膜收缩蛋白联合缺乏最常见。上述膜蛋白异常可导致膜骨架与膜之间的垂直方向相互作用减弱,从而使膜脂质双层变得不稳定,部分脂质以出芽形式形成囊泡而丢失,红细胞膜表面积减少,最终使红细胞形成小球形。另外,HS红细胞(特别是经过脾脏的红细胞)都有一定程度的脱水和对单价离子通透性异常,这可能也与膜骨架缺陷有关。 由于球形细胞内容积储备很低,其变形性能因而降低,难于通过直径比其本身小得多的脾微循环而阻留...[详细]
1.病理生理 (1)阳离子含量的改变与渗透性:红细胞内外物质交换需要通过细胞膜,红细胞内外无机离子、糖等的浓度差别很大,它们的转运都有各自的机制。正常红细胞通过Na/K泵维持细胞内Na/K正常比例,Na/K泵每作用1次即有3个Na泵出细胞外,2个K泵入细胞内,使红细胞内呈高钾低钠状态。而HS红细胞,特别是从脾脏收集的红细胞存在脱水异常和对单价离子的通透性异常,推测是骨架蛋白缺乏的结果。钾和水选择性丢失的途径被激活引起细胞的脱水异常,如脾脏的相对低pH值和氧化作用的损伤以及红细胞在脾脏与巨噬细胞接触产生氧自由基可刺激K/Cl-联合输送器。此外,在HS红细胞,调节细胞内钠和钾含量的Na/K泵的活动是亢进的。因为每2个原子的钾转运进入细胞内,3个钠原子从细胞内被挤出,泵的功能亢进将导致红细胞脱水以免红细胞肿胀、破坏。蛋白4.2缺乏的HS红细胞有阴离子输送增加,而血影蛋白、锚蛋白或带3缺乏的HS红细胞阴离子输送正常或输送减少。 (2)非变形性球形红细胞在脾脏的滞留:脾脏在HS发病中病理生理机制的重要性众所周知。脾脏选择性破坏HS红细胞有两个因素:一是HS红细胞变形性不良,二是脾脏血管系统的独特解剖构造充当“微循环过滤器”的作用。 由于表面物质缺失引起红细胞表面积与体积的比值减少,从而导致红细胞变形性差是发病中的主要因素。正常的盘状细胞具有丰富的表面,允许红细胞变形并通过狭窄的微循环信道,而HS红细胞缺少这部分可变形的额外表面。变形性不良可由于细胞的脱水更进一步加重。 红细胞在脾脏滞留的主要部位是脾脏窦孔的壁,来自脾脏红髓脾索的血液进入静脉循环。在大鼠的脾脏,孔的长与宽分别为2~3μm和0.2~0.5μm,约为红细胞直径的一半。脾标本的电子纤维照片显示只有非常少量的HS红细胞穿过此部位。因此,在切除的脾脏可以观察到解剖部位的非变形性球形红细胞堆积于红髓,使红髓充血变粗。 (3)脾脏对红细胞的调节与破坏:HS红细胞由于表面区域缺失和细胞密度的增加,一旦经脾脏扣留将经受附加的损伤,在脾切除时有红细胞移出脾脏就是证据。这些经脾脏处理过的红细胞重返血液循环,可以通过渗透脆性检测出这部分细胞群。脾切除后,这些红细胞群消失。 早期通过模拟脾脏条件(包括低pH值、隔离的红细胞可以与网状内皮系统接触等)进行HS红细胞的体外培养研究显示,糖缺失和接踵而来的细胞内ATP的缺乏并非HS红细胞在脾脏破坏的原因。脾脏条件的影响显示出的是累积的损伤。HS红细胞在脾索停留的时间平均为10~100min,只有1%~10%流经脾脏的血液是暂时滞留于脾脏并充满脾索,其余的90%血液迅速流入静脉循环。 虽然HS红细胞主要在脾脏扣留和破坏,但HS细胞也在其他外周的器官被破坏。HS红细胞表面改变触发网状内皮系统吞噬作用的机制尚不清楚。一个途径可能是脂质双分子层结构内磷脂的破坏,导致磷脂酰丝氨酸外侧暴露,促进红细胞附着于网状内皮系统,使其在脾脏以外的其他器官被吞噬破坏。虽然磷脂在两个脂质双层内的分布在绝大多数HS患者是正常的,但在一些严重的HS患者有磷脂分布的异常改变。还有一种猜测,经脾脏处理过的末期HS红细胞具有磷脂不均匀的发生。 2.分子机制 正常红细胞的膜是有非酯化胆固醇和糖脂插入的不对称性磷脂双层结构。膜的外层为胆碱磷脂(磷脂酰胆碱也称卵磷脂和鞘磷脂),内层为氨基酸磷脂(磷脂酰氨基乙醇和磷脂酰丝氨酸)。红细胞膜也含有不对称的蛋白成分。所有的糖蛋白暴露于膜的外侧表面,具有红细胞抗原和受体或转运蛋白。这个整体的膜蛋白穿透或跨越脂质双层,与疏水脂质的核心部位相互作用并且紧紧地束缚红细胞膜。一个独立的蛋白网络与整体膜蛋白和脂质双层形成垂直和水平相互作用的膜骨架。膜骨架包括血影蛋白(或称收缩蛋白,又分为α和β spectrin)、锚蛋白(ankyrin)、蛋白4.1、蛋白4.2和肌动蛋白(图1)。HS的分子改变具有异质性。通过聚丙烯酰胺凝胶电泳(PAGE)密度梯度测定法定量分离膜蛋白,HS被分为以下5种亚型:单一血影蛋白部分缺乏、血影蛋白与锚蛋白连接部分缺乏、带3部分缺乏、蛋白4.2缺乏和其他少见的共同缺乏。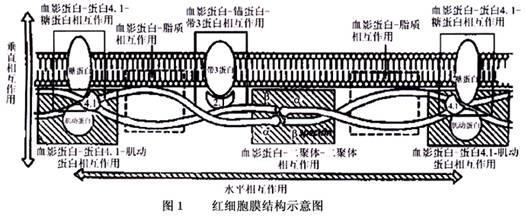 (1)单一的血影蛋白部分缺乏:单一血影蛋白部分缺乏包括α-血影蛋白和β-血影蛋白。大量文献证实在血影蛋白缺乏的显性遗传HS患者有β-血影蛋白基因(SPTB)突变存在。但有一种例外,β-血影蛋白Houston在一些家族被证明是一种移码突变,这些突变是局限的,是独特的个体家族,并且可能与β-血影蛋白mRNA减少的积累有关。β-血影蛋白Kissimmee是一种局限在与蛋白4.1交互作用的β-血影蛋白的高度保守区域的点突变,是一种限制蛋白4.1与血影蛋白到肌动蛋白连接键的功能障碍。因此,通过还原剂处理循环中的红细胞来增强其限制功能。这些红细胞富含还原型谷胱甘肽,血影蛋白/蛋白4.1限制性减低是非功能性的表现。 单一血影蛋白缺乏的非显性遗传HS患者,属α-血影蛋白的缺乏。在正常的红细胞,α-血影蛋白合成量大大超过β-血影蛋白,α-血影蛋白基因(SPTA1)突变导致α-血影蛋白合成减少。由于α-血影蛋白超过β-血影蛋白的合成,因而,在膜内有一个正常数量的血影蛋白异二聚体组合,因此,一个正常的α-血影蛋白和一个有缺陷的α-血影蛋白等位基因结合的个体可无症状。纯合子或复合杂合子的α-血影蛋白缺乏HS个体,将是严重型的HS患者。Wichterle等报道1例复合杂合子的α-血影蛋白缺乏严重的HS病例,具有两个不同的α-血影蛋白基因缺乏,在一个等位基因上,有一个与上游间插序列突变(αLEPRA)有关的剪切缺失;在另一个等位基因上有另外一个基因突变,即aPRAGUE。αLEPRA 等位基因产生比正常的等位基因少6倍的纠正剪切的α血影蛋白转录物。更进一步的研究显示,许多非显性遗传的血影蛋白缺乏HS,αLEPRA与αBug Hill(在αⅡ结构域部位有一个氨基酸取代了结构域)的连接是失平衡的。因此αLEPRA等位基因与其他α-收缩蛋白缺乏的杂合个体出现,导致明显的血影蛋白缺乏的球形红细胞增多性溶血性贫血。 利用脉冲标记的BFU-E研究显示,在一些致死性或接近致死性的与血影蛋白严重缺乏(约占正常成分的26%)的HS,其α-血影蛋白的合成显著减低。尽管这些缺陷的分子基础尚不清楚,但有母亲是轻度显性遗传的HS和有渗透脆性轻度增加而血液学表现正常的父亲的家族史,提示有至少两个基因缺陷的简单杂合子的可能。 (2)血影蛋白与锚蛋白的结合部分缺乏:血影蛋白与锚蛋白结合部分缺乏的生物化学表现最先在1988年由Coetzer等提出。锚蛋白代表血影蛋白在膜上的主要连接部位,因此,尽管血影蛋白的合成正[收起]
1.病理生理 (1)阳离子含量的改变与渗透性:红细胞内外物质交换需要通过细胞膜,红细胞内外无机离子、糖等的浓度差别很大,它们的转运都有各自的机制。正常红细胞通过Na/K泵维持细胞内Na/K正常比例,Na/K泵每作用1次即有3个Na泵出细胞外,2个K泵入细胞内,使红细胞内呈高钾低钠状态。而HS红细胞,特别是从脾脏收集的红细胞存在脱水异常和对单价离子的通透性异常,推测是骨架蛋白缺乏的结果。钾和水选择性丢失的途径被激活引起细胞的脱水异常,如脾脏的相对低pH值和氧化作用的损伤以及红细胞在脾脏与巨噬细胞接触产生氧自由基可刺激K/Cl-联合输送器。此外,在HS红细胞,调节细胞内钠和钾含量的Na...[详细]
(1)单一的血影蛋白部分缺乏:单一血影蛋白部分缺乏包括α-血影蛋白和β-血影蛋白。大量文献证实在血影蛋白缺乏的显性遗传HS患者有β-血影蛋白基因(SPTB)突变存在。但有一种例外,β-血影蛋白Houston在一些家族被证明是一种移码突变,这些突变是局限的,是独特的个体家族,并且可能与β-血影蛋白mRNA减少的积累有关。β-血影蛋白Kissimmee是一种局限在与蛋白4.1交互作用的β-血影蛋白的高度保守区域的点突变,是一种限制蛋白4.1与血影蛋白到肌动蛋白连接键的功能障碍。因此,通过还原剂处理循环中的红细胞来增强其限制功能。这些红细胞富含还原型谷胱甘肽,血影蛋白/蛋白4.1限制性减低是非功能性的表现。 单一血影蛋白缺乏的非显性遗传HS患者,属α-血影蛋白的缺乏。在正常的红细胞,α-血影蛋白合成量大大超过β-血影蛋白,α-血影蛋白基因(SPTA1)突变导致α-血影蛋白合成减少。由于α-血影蛋白超过β-血影蛋白的合成,因而,在膜内有一个正常数量的血影蛋白异二聚体组合,因此,一个正常的α-血影蛋白和一个有缺陷的α-血影蛋白等位基因结合的个体可无症状。纯合子或复合杂合子的α-血影蛋白缺乏HS个体,将是严重型的HS患者。Wichterle等报道1例复合杂合子的α-血影蛋白缺乏严重的HS病例,具有两个不同的α-血影蛋白基因缺乏,在一个等位基因上,有一个与上游间插序列突变(αLEPRA)有关的剪切缺失;在另一个等位基因上有另外一个基因突变,即aPRAGUE。αLEPRA 等位基因产生比正常的等位基因少6倍的纠正剪切的α血影蛋白转录物。更进一步的研究显示,许多非显性遗传的血影蛋白缺乏HS,αLEPRA与αBug Hill(在αⅡ结构域部位有一个氨基酸取代了结构域)的连接是失平衡的。因此αLEPRA等位基因与其他α-收缩蛋白缺乏的杂合个体出现,导致明显的血影蛋白缺乏的球形红细胞增多性溶血性贫血。 利用脉冲标记的BFU-E研究显示,在一些致死性或接近致死性的与血影蛋白严重缺乏(约占正常成分的26%)的HS,其α-血影蛋白的合成显著减低。尽管这些缺陷的分子基础尚不清楚,但有母亲是轻度显性遗传的HS和有渗透脆性轻度增加而血液学表现正常的父亲的家族史,提示有至少两个基因缺陷的简单杂合子的可能。 (2)血影蛋白与锚蛋白的结合部分缺乏:血影蛋白与锚蛋白结合部分缺乏的生物化学表现最先在1988年由Coetzer等提出。锚蛋白代表血影蛋白在膜上的主要连接部位,因此,尽管血影蛋白的合成正[收起]
1.病理生理 (1)阳离子含量的改变与渗透性:红细胞内外物质交换需要通过细胞膜,红细胞内外无机离子、糖等的浓度差别很大,它们的转运都有各自的机制。正常红细胞通过Na/K泵维持细胞内Na/K正常比例,Na/K泵每作用1次即有3个Na泵出细胞外,2个K泵入细胞内,使红细胞内呈高钾低钠状态。而HS红细胞,特别是从脾脏收集的红细胞存在脱水异常和对单价离子的通透性异常,推测是骨架蛋白缺乏的结果。钾和水选择性丢失的途径被激活引起细胞的脱水异常,如脾脏的相对低pH值和氧化作用的损伤以及红细胞在脾脏与巨噬细胞接触产生氧自由基可刺激K/Cl-联合输送器。此外,在HS红细胞,调节细胞内钠和钾含量的Na...[详细]
临床表现有显著异质性。起病年龄和病情轻重差异很大,HS多见于幼儿或儿童期,从无症状到危及生命的贫血,重者于新生儿或婴儿期起病。北京儿童医院收治的170例中,5岁以内发病139例,占82%,其中一半在1岁以内发病。不同家族之间,临床表现的轻重可有很大差别,同一家族的不同患者,病情轻重常较一致。 根据临床表现,可将HS分为4型:无症状携带者、轻型HS、典型HS和重型HS。多数患儿为显性遗传,临床表现为轻中度贫血;极少数患儿为隐性遗传的纯合子或等位基因都发生突变,临床表现为重型HS。 贫血、黄疸和肝脾肿大是HS最常见的临床表现,三者或同时存在,或单独发生。北京儿童医院收治的170例HS中,贫血169例(99%),黄疸133例(78%),肝大155例(91%),脾大168例(99%),构成本病的四大表现。 大多数HS有轻中度贫血、中度脾肿大和间歇性黄疸。少数(约25%)HS症状轻微,虽然有溶血,但由于骨髓红系代偿性增生,一般无贫血,无或仅有轻微黄疸,无或有轻度脾肿大。这类病人只在进行家族调查或某种诱因导致红细胞破坏加重时才被发现。 最常见的诱因是感染,剧烈体力活动也可加重溶血。极少数HS可发生危及生命的溶血,需要定期输血,生长发育也可受到影响。长期明显贫血者,由于骨髓增生、骨髓腔变宽,使额骨和颞骨突起。 新生儿期起病者,黄疸的发生率约50%,常于出生后48h内出现,并可因高胆红素血症而发生胆红素脑病。新生儿期后,黄疸大多很轻,呈间歇性发作,劳累、感染均可诱发或加重黄疸。[收起]
临床表现有显著异质性。起病年龄和病情轻重差异很大,HS多见于幼儿或儿童期,从无症状到危及生命的贫血,重者于新生儿或婴儿期起病。北京儿童医院收治的170例中,5岁以内发病139例,占82%,其中一半在1岁以内发病。不同家族之间,临床表现的轻重可有很大差别,同一家族的不同患者,病情轻重常较一致。 根据临床表现,可将HS分为4型:无症状携带者、轻型HS、典型HS和重型HS。多数患儿为显性遗传,临床表现为轻中度贫血;极少数患儿为隐性遗传的纯合子或等位基因都发生突变,临床表现为重型HS。 贫血、黄疸和肝脾肿大是HS最常见的临床表现,三者或同时存在,或单独发生。北京儿童医院收治的170...[详细]
在疾病的任何阶段均可能发生贫血危象: 1.溶血危象 劳累、急性感染、受冷等因素可诱发急性溶血而发生“溶血危象”,多与吞噬细胞功能一过性增强有关,常呈自限性。 2.再生障碍危象 较少见,症状重,可危及生命,常需要输血。主要由细小病毒B19感染引起。此病毒可侵入红系祖细胞而抑制其增殖和分化。 少数年长儿患者可并发胆石症(10岁以下的发生率约5%),重者可并发阵发性胆绞痛和阻塞性黄疸。还有少数患儿可并发下肢复发性溃疡,这可能与红细胞变形性降低、局部血流淤滞有关。
1.血象 轻、中度或重度贫血均可发生,也可无贫血。网织红细胞增高,为5%~20%,最低2%,也有高过20%者。白细胞数正常或稍增,在溶血危象时可增高。血小板数正常。再生障碍危象时,贫血加重,甚至全血细胞减少,网织红细胞也减少。 红细胞形态:血涂片镜检可见小球形红细胞(图3),这些细胞数目多少不一,一般占红细胞的20%~30%,亦有仅占1%~2%者。其特征是细胞直径小(6.2~7.01μm),厚度增大,为2.2~3.4μm(正常为1.9~2.0μm),胞体小而染色深,无中央淡染区及双凹盘状。小球形红细胞仅限于成熟红细胞,有核红细胞和网织红细胞形态正常。在重型HS,血涂片除可见到大量小球形红细胞外,还可见到许多棘形红细胞。MCV仅轻度减小,MCHC增高。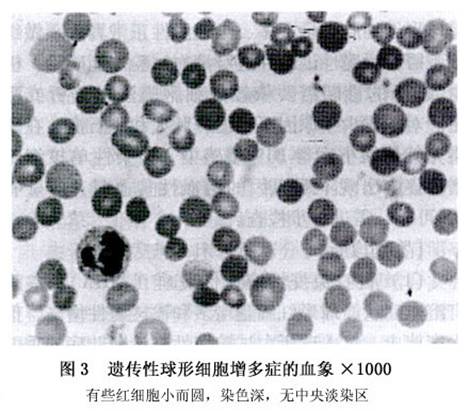 2.骨髓象 红细胞系统增生极度活跃,以中晚幼红细胞居多。在再生障碍危象时,红细胞系统增生低下,有核红细胞减少。 3.红细胞渗透脆性试验 是确诊本症的主要方法。绝大多数病例红细胞渗透脆性增高,增高的程度与球形细胞的数量成正比。球形红细胞数量很少者,红细胞渗透脆性试验也可以正常,须将红细胞在37℃孵育24h后才能发现其渗透脆性增高。红细胞机械脆性增高。再生障碍危象和合并铁缺乏时,红细胞渗透脆性可相应降低。 4.红细胞自身溶血及自溶纠正试验 48h的溶血度明显增加,可以达到10%~50%(正常5%),加入葡萄糖或ATP可不完全纠正。 5.酸化甘油溶解试验(AGLT50) 正常人红细胞AGLT50约为1800s,重症HS患者AGLT50可在150s内。该法操作简单,适用于诊断和筛查。 6.红细胞膜蛋白定性分析 可采用SDS-PAGE对膜蛋白定性分析,80%以上的HS可发现异常,结合免疫印迹法,可提高可信性。还可采用放射免疫法或ELISA法直接对每个红细胞的膜蛋白进行定量分析。 7.其他 血清未结合胆红素增高,尿胆原正常或增高,粪胆原增高。51Cr标记测定红细胞寿命缩短,其半衰期(T1/2)为8~18天。血清结合珠蛋白下降,乳酸脱氢酶增高。Coombs试验阴性。血清叶酸水平一般降低。[收起]
1.血象 轻、中度或重度贫血均可发生,也可无贫血。网织红细胞增高,为5%~20%,最低2%,也有高过20%者。白细胞数正常或稍增,在溶血危象时可增高。血小板数正常。再生障碍危象时,贫血加重,甚至全血细胞减少,网织红细胞也减少。 红细胞形态:血涂片镜检可见小球形红细胞(图3),这些细胞数目多少不一,一般占红细胞的20%~30%,亦有仅占1%~2%者。其特征是细胞直径小(6.2~7.01μm),厚度增大,为2.2~3.4μm(正常为1.9~2.0μm),胞体小而染色深,无中央淡染区及双凹盘状。小球形红细胞仅限于成熟红细胞,有核红细胞和网织红细胞形态正常。在重型HS,血涂片除可见到大量小...[详细]
2.骨髓象 红细胞系统增生极度活跃,以中晚幼红细胞居多。在再生障碍危象时,红细胞系统增生低下,有核红细胞减少。 3.红细胞渗透脆性试验 是确诊本症的主要方法。绝大多数病例红细胞渗透脆性增高,增高的程度与球形细胞的数量成正比。球形红细胞数量很少者,红细胞渗透脆性试验也可以正常,须将红细胞在37℃孵育24h后才能发现其渗透脆性增高。红细胞机械脆性增高。再生障碍危象和合并铁缺乏时,红细胞渗透脆性可相应降低。 4.红细胞自身溶血及自溶纠正试验 48h的溶血度明显增加,可以达到10%~50%(正常5%),加入葡萄糖或ATP可不完全纠正。 5.酸化甘油溶解试验(AGLT50) 正常人红细胞AGLT50约为1800s,重症HS患者AGLT50可在150s内。该法操作简单,适用于诊断和筛查。 6.红细胞膜蛋白定性分析 可采用SDS-PAGE对膜蛋白定性分析,80%以上的HS可发现异常,结合免疫印迹法,可提高可信性。还可采用放射免疫法或ELISA法直接对每个红细胞的膜蛋白进行定量分析。 7.其他 血清未结合胆红素增高,尿胆原正常或增高,粪胆原增高。51Cr标记测定红细胞寿命缩短,其半衰期(T1/2)为8~18天。血清结合珠蛋白下降,乳酸脱氢酶增高。Coombs试验阴性。血清叶酸水平一般降低。[收起]
1.血象 轻、中度或重度贫血均可发生,也可无贫血。网织红细胞增高,为5%~20%,最低2%,也有高过20%者。白细胞数正常或稍增,在溶血危象时可增高。血小板数正常。再生障碍危象时,贫血加重,甚至全血细胞减少,网织红细胞也减少。 红细胞形态:血涂片镜检可见小球形红细胞(图3),这些细胞数目多少不一,一般占红细胞的20%~30%,亦有仅占1%~2%者。其特征是细胞直径小(6.2~7.01μm),厚度增大,为2.2~3.4μm(正常为1.9~2.0μm),胞体小而染色深,无中央淡染区及双凹盘状。小球形红细胞仅限于成熟红细胞,有核红细胞和网织红细胞形态正常。在重型HS,血涂片除可见到大量小...[详细]
常规做影像学检查,如胸片、B超,注意有无肺部感染,胆石和肝脾肿大等。
典型病例可根据黄疸、贫血、脾肿大、球形红细胞增多,网织红细胞增多,红细胞脆性增高和阳性家族史等做出诊断。轻型病例,特别是球形红细胞数量不多、渗透脆性正常者,须做红细胞孵育后脆性试验和自身溶血试验才能确诊。极少数HS的诊断需要依赖红细胞膜蛋白分析或测定。对于青少年原因不明的脾肿大和胆石症,在感染尤其是细小病毒B19型感染、传染性单核细胞增多症中出现不明原因的溶血性贫血时,应疑有HS可能,需进一步检查。
血红蛋白<70g/L时,应适当输注红细胞,以改善贫血。脾切除是治疗本症的根本办法,凡确诊者都应进行脾切除术治疗。极轻症患者,可将手术时间推迟,并追踪观察病情变化,以决定是否需行手术。年幼儿因免疫功能尚未完善,术后患暴发性感染,特别是肺炎双球菌、大肠埃希杆菌的感染机会较多,因此小儿手术年龄以5岁以上为宜。对重症患儿,如频繁发作溶血或再障危象,手术年龄亦可适当提前,但应禁忌在1岁以内进行。小年龄手术者术后应以苄星青霉素(长效青霉素)注射半年到1年。脾切除后红细胞膜缺陷和球形红细胞依然存在,但由于除去了主要破坏血细胞的场所,红细胞寿命得以延长,使贫血获得纠正,黄疸迅速消退。 脾切除术过程中应注意寻找副脾,特别注意脾门、脾韧带、大网膜等好发部位。如有副脾,应一并切除。 为了降低脾切除术后并发症的发生率,国外正尝试改进手术方式(包括进行部分脾切除术),但疗效及优越性有待进一步确定。部分脾动脉栓塞术和骨髓移植治疗HS尚在研究中。 如发生贫血危象,应予输血、补液和控制感染。本病在溶血过程中,对叶酸的需要量增加,应注意补充。 新生儿期发病者,主要针对高胆红素血症进行治疗。[收起]
血红蛋白<70g/L时,应适当输注红细胞,以改善贫血。脾切除是治疗本症的根本办法,凡确诊者都应进行脾切除术治疗。极轻症患者,可将手术时间推迟,并追踪观察病情变化,以决定是否需行手术。年幼儿因免疫功能尚未完善,术后患暴发性感染,特别是肺炎双球菌、大肠埃希杆菌的感染机会较多,因此小儿手术年龄以5岁以上为宜。对重症患儿,如频繁发作溶血或再障危象,手术年龄亦可适当提前,但应禁忌在1岁以内进行。小年龄手术者术后应以苄星青霉素(长效青霉素)注射半年到1年。脾切除后红细胞膜缺陷和球形红细胞依然存在,但由于除去了主要破坏血细胞的场所,红细胞寿命得以延长,使贫血获得纠正,黄疸迅速消退。 脾切除术过程中...[详细]
在新生儿或婴儿期起病者,因溶血危象发作较频,其预后较差,可因严重贫血并发心力衰竭而死亡。起病较晚者因慢性贫血可致发育迟缓。轻症或无症状者不影响生长发育,预后一般较好。极少数可以死于贫血危象或脾切除后并发症。
本病属常染色体显性遗传性疾病,预防措施同遗传性疾病,预防应从孕前贯穿至产前。 婚前体检在预防出生缺陷中起到积极的作用,作用大小取决于检查项目和内容,主要包括血清学检查(如乙肝病毒、梅毒螺旋体、艾滋病病毒)、生殖系统检查(如筛查宫颈炎症)、普通体检(如血压、心电图)以及询问疾病家族史、个人既往病史等,做好遗传病咨询工作。 孕妇尽可能避免危害因素,包括远离烟雾、酒精、药物、辐射、农药、噪音、挥发性有害气体、有毒有害重金属等。在妊娠期产前保健的过程中需要进行系统的出生缺陷筛查,包括定期的超声检查、血清学筛查等,必要时还要进行染色体检查。 一旦出现异常结果,需要明确是否可治疗,预后如何等等。采取切实可行的诊治措施。[收起]
本病属常染色体显性遗传性疾病,预防措施同遗传性疾病,预防应从孕前贯穿至产前。 婚前体检在预防出生缺陷中起到积极的作用,作用大小取决于检查项目和内容,主要包括血清学检查(如乙肝病毒、梅毒螺旋体、艾滋病病毒)、生殖系统检查(如筛查宫颈炎症)、普通体检(如血压、心电图)以及询问疾病家族史、个人既往病史等,做好遗传病咨询工作。 孕妇尽可能避免危害因素,包括远离烟雾、酒精、药物、辐射、农药、噪音、挥发性有害气体、有毒有害重金属等。在妊娠期产前保健的过程中需要进行系统的出生缺陷筛查,包括定期的超声检查、血清学筛查等,必要时还要进行染色体检查。 一旦出现异常结果,需要明确是否...[详细]
 浙公网安备
33010902000463号
浙公网安备
33010902000463号