概述
半乳糖血症系半乳糖-1-磷酸尿苷酸转移酶(galactose-1-phosphate uridyl transferase,Gal-1-PUT)缺乏所引起的先天性代谢紊乱性疾病。 半乳糖被吸收后,只能经由一条途径进行代谢,即首先在半乳糖激酶的催化下,半乳糖被磷酸化成半乳糖-1-磷酸。此化合物在半乳糖-1-磷酸尿苷转移酶的催化下,与二磷酸尿苷葡萄糖(UDPG)反应,生成二磷酸尿苷半乳糖(UDPGal) 和1-磷酸葡萄糖;UDPGal经二磷酸尿苷半乳糖异构酶作用,转化为UDPa。新的UDPG又可与另一分子半乳糖-1-磷酸反应而产生1-磷酸葡萄糖。半乳糖分解代谢中的这3种酶(激酶、转移酶和...[详细]
病因
本病为常染色体隐性遗传病。病儿红细胞和肝细胞中缺乏Gal-1-PUT致使半乳糖-1-磷酸转变成葡萄糖-1-磷酸的过程受阻,导致半乳糖和半乳糖-1-磷酸大量聚积在血流和组织内。
发病机制
1.发病机制 半乳糖-1-磷酸尿苷酰转移酶的编码基因(GLAT)位于9p13~p21,它的缺陷即导致半乳糖、半乳糖-1-磷酸和半乳糖代谢旁路生成的半乳糖醇在各种组织中积累。1-磷酸半乳糖具细胞毒性,对糖代谢途径中的多种酶有抑制作用,特别是葡糖磷酸变位酶的作用被阻抑后不能使1-磷酸葡萄糖转化为6-磷酸葡萄糖,阻断了糖原的分解过程;高浓度的1-磷酸半乳糖还抑制葡萄糖异生过程,因而在临床上呈现低血糖症状。半乳糖进入晶体后即被醛糖还原酶(aldose reductase)还原成为半乳糖醇,沉积在晶体中造成晶体内渗透压增高、含水量增加、氨基酸转运和蛋白合成降低等代谢异常,最终形成白内障。本型患儿的肝、...[详细]
临床表现
典型者在围生期即发病,常在喂给乳类后数天即出现呕吐、拒食、体重不增和嗜睡等症状,继而呈现黄疸和肝脏肿大。若不能及时诊断而继续喂给乳类,将导致病情进一步恶化,在2~5周内发生腹水、肝功能衰竭、出血等终末期症状。如用裂隙灯检查,在发病早期即可发现晶体白内障形成。约30%~50%患儿在病程第1周左右并发大肠埃希杆菌败血症,使病情更加严重。未经及时诊断和治疗的患儿大多在新生儿期内夭折。少数患儿症状可较轻微,仅在进食乳类后出现轻度的消化道症状,但如继续使用乳类食物则在幼婴儿期逐渐呈现生长迟缓、智能发育落后、肝硬化和白内障等征象。
并发症
黄疸和肝脏肿大、肝硬化,发生腹水,肝功能衰竭,出血,并发大肠埃希杆菌败血症,生长迟缓、智能发育落后等。
实验室检查
早期正确诊断需依赖实验室检测。 1.新生儿期筛查 通过对新生儿进行群体筛查不仅可以达到早期诊断和治疗的目的,还可以为遗传咨询和计划生育提供资料。大多数筛查中心都选用两种方法: (1)Beutler试验:用于检测血滴纸片的半乳糖-1-磷酸尿酰转移酶活性,其缺点是假阳性率过高。 (2)Paigen试验:是用于检测血滴纸片半乳糖和半乳糖-1-磷酸的半定量方法,优点是很少假阳性,并且3种酶缺陷都可被检出。应用双质谱联用仪(tandem MS)进行筛查尤为便捷、正确。 2.尿液中还原糖测定 对有疑似症状的患儿都必须及时检查其尿中是否含有还原糖。尿液中可...[详细]
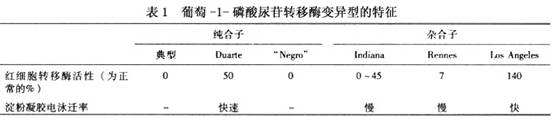 4.其他 必要时应检测肝功能、凝血机制、血糖、血电解质和血、尿培养等项目以利诊断。
4.其他 必要时应检测肝功能、凝血机制、血糖、血电解质和血、尿培养等项目以利诊断。其他辅助检查
常规做X线、B超等检查,可发现肝脏肿大、肝硬化、脾大、腹水等。裂隙灯检查,可发现白内障,尚可发现眼底病变,如视网膜剥离、眼球内出血等。脑电图检查可发现异常波形。
诊断
诊断主要根据临床症状及相关酶活性测定确诊。尿中葡萄糖水平正常而班氏试验阳性者应疑为半乳糖血症,结合红细胞内半乳糖代谢酶缺乏通常可确诊。如果产前怀疑胎儿可能有半乳糖血症,可通过羊膜穿刺术进行产前诊断,或出生时取脐带血检查红细胞内的酶活性。值得注意的是,通过羊膜穿刺术并不能了解胎儿的大脑发育是否已经受到损害。出生后2个月内的新生儿,进行半乳糖血症诊断时应排除新生儿暂时性半乳糖血症(transient neonatal galactosemia)可能,这是因为肝脏功能尚未完全成熟所致,其特点是血中半乳糖轻度升高、血α-胎儿蛋白(AFP)升高,数月后可自动恢复正常,患儿尿中无半乳糖醇和半乳糖酸,可与半...[详细]
治疗
1.限制乳类 立刻停用乳类,改用豆浆、米粉等,并辅以维生素、脂肪等营养必需物质。豆浆中虽含有能分解出半乳糖的蜜三糖(raffinose)和水苏糖(stachyose),但不能被人体肠道吸收,故无碍于治疗。通常在限制乳类3~4天后即可见临床症状改善,肝功能在1周后好转。 在患儿开始摄食辅助食物以后,必须避免一切可能含有奶类的食品和某些含有乳糖的水果、蔬菜如西瓜、西红柿等。 2.支持治疗 静脉输给葡萄糖、新鲜血浆,注意补充电解质。 3.抗生素 对合并败血症的患儿应采用适当的抗生素,并给予积极支持治疗。
预后
患儿的预后取决于能否得到早期诊断和治疗。未经正确治疗者大都在新生儿期死亡,平均寿命约为6周,即便幸免,日后亦遗留智能发育障碍。获得早期确诊的患儿生长发育大多正常,但多数在成年后可有学习障碍、语言困难或行为异常等问题。女性患儿在年长后几乎都发生性腺功能不足,原因尚不甚清楚。 开始控制饮食的时间越早,则患儿的预后越好。尽管患儿的智商可在正常范围之内,但学习成绩仍比不上正常儿童。由于患儿体内半乳糖代谢酶的缺乏并不会随年龄增长而逐渐改善,因此需终身进行饮食控制。不能坚持饮食控制者,可发生不同程度的智力低下、生长障碍及白内障。
预防
发病机制尚未完全清楚,预防措施参照遗传性疾病进行。 由于已证明病儿皮肤成纤维细胞有Gal-1-PUT酶异常,因此通过培养羊水细胞的酶分析显示Gal-1-PUT活性减低,可做出产前诊断。Nadler等已用此法进行产前诊断,但由于有简便易行的新生儿筛选方法,而且若能早期发现并控制饮食,病儿能正常发育,故对是否应做产前诊断尚有争议。 患者在妊娠期间限制牛奶和奶制品的摄入,这样既可减轻母亲的病情又可预防婴儿发生先天性白内障。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号