丹迪—沃克综合征是怎么个情况,咋处理?
2017年02月20日 14460人阅读 返回文章列表
【别名】
丹迪—沃克畸形、先天性第四脑室中侧孔闭锁
【概述】
丹迪-沃克综合征(Dandy-Walker’s syndrome)又称丹迪-沃克畸形,或先天性第四脑室中侧孔闭锁,系小脑蚓部部分或全部缺失,第四脑室囊性扩张,常合并脑积水的罕见疾病。约占儿童先天性脑积水的2%~4%。
【病因与发病机制】
本病病因和发病机制不完全清楚,可能是遗传、感染、代谢等多种因素引起的胚胎发生学异常。大多数为散发。少数与遗传有关,多为AR遗传,部分为AD遗传和X连锁遗传。
【病理】
病理可见第四脑室囊状或憩室样扩大,小脑蚓部发育不全或缺失;囊肿壁由后髓帆组成,包括室管膜、胶质、小脑、软脑膜及蛛网膜等组织。小脑被推向外上方。第四脑室正中孔大多闭锁,但约50%的患者一侧或两侧侧孔仍然开放。窦汇、横窦和小脑幕位置抬高,常并有脑室系统扩张和脑积水。
【诊断要点】
临床表现
本病以婴幼儿多见,约60%见于2岁内,常因运动发育迟缓就医,成人少见,往往以颅高压就诊。约80%的患儿在1岁内有脑积水。国外文献提及本病女性占53.50%~65%,国内资料男女之比为1.48∶1。临床表现为脑积水征:头颅进行性增大、前囟门膨隆,枕骨区突出,头颅叩诊有“破壶音”,“落日眼”征,兴奋性增高,头痛、呕吐等;小脑征:眼球水平震颤、共济失调、步态不稳;其他神经系统症状:50%以上有运动发育迟缓、智力低下,还有痉挛性瘫痪、癫癎发作等。
2/3以上的患儿可合并其他中枢神经系统畸形或器官畸形,如胼胝体发育不全、脑膜膨出、神经元移行异常、脂肪瘤、畸胎瘤等;骨骼畸形占25%以上,包括多指、并指、颅裂、颈椎融合畸形等;心血管异常为房室间隔缺损、动脉导管未闭、脑血管畸形等。国内王宾民等(1997)报道1例18岁的丹迪-沃克综合征患者,伴有脑膜膨出、颈椎融合、Wildervanck综合征(颈椎-眼-内耳畸形)等。
影像学检查
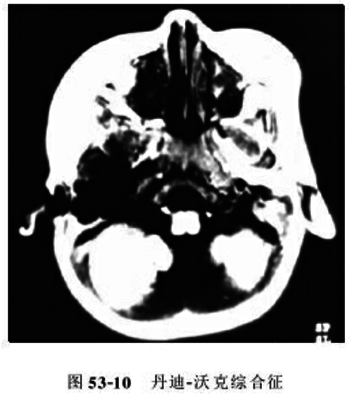
颅骨X线检查可有颅骨变薄、骨缝分离、蝶鞍扩大、前后床突骨质吸收、颅后窝扩大。CT和MRI检查可显示小脑蚓部部分或完全缺失;第4脑室呈囊袋状、扇形或三角形,从缺如的小脑蚓部向后上方扩张,占据颅后窝或与巨大囊肿相通;小脑半球向两侧分离并被推向外上方,且有不同程度退缩变小;可有脑干前移、幕上脑室系统对称性扩大(图)。MRI矢状位示小脑幕、窦汇和横窦被抬高。
鉴别诊断
根据婴幼儿出现脑积水征等上述临床表现,头颅CT或MRI检查有脑积水、小脑蚓部发育不全、颅后窝囊肿等改变可作出诊断。应与以下疾病鉴别。①巨型枕大池:可能是解剖变异,其第四脑室及小脑蚓部发育正常;②颅后窝巨大蛛网膜囊肿:可引起第四脑室向前移位,幕上脑积水,小脑幕抬高,但不与脑室系统相通,小脑蚓部发育正常。
【治疗概述】
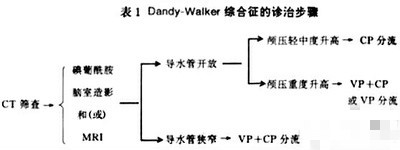
本病主要为手术治疗,目的是控制颅内压增高,包括切除囊肿、在第四脑室和蛛网膜下腔之间建立交通、侧脑室分流。B超可行产前诊断,选择性终止妊娠,减少或杜绝患儿出生。
 浙公网安备
33010902000463号
浙公网安备
33010902000463号